
Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The H3K4me3-Mediated GCR Does Not Induce Up-Regulation of HBG1/2 Expression in the Context of HBB-KO
2.2. Chromatin Accessibility Analysis Suggests That Erythroid Differentiation Is Enhanced in HBB-KO Cells
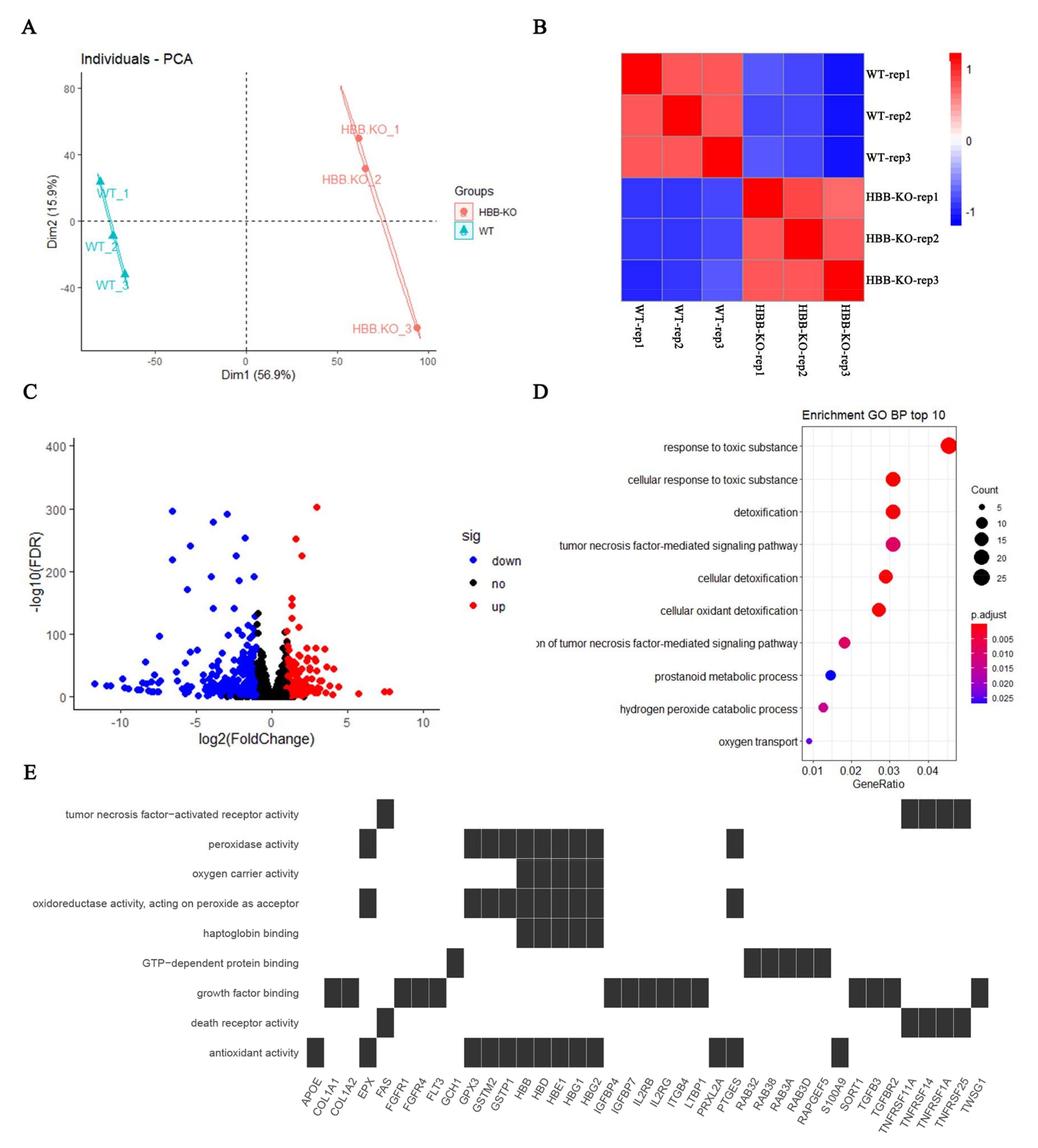
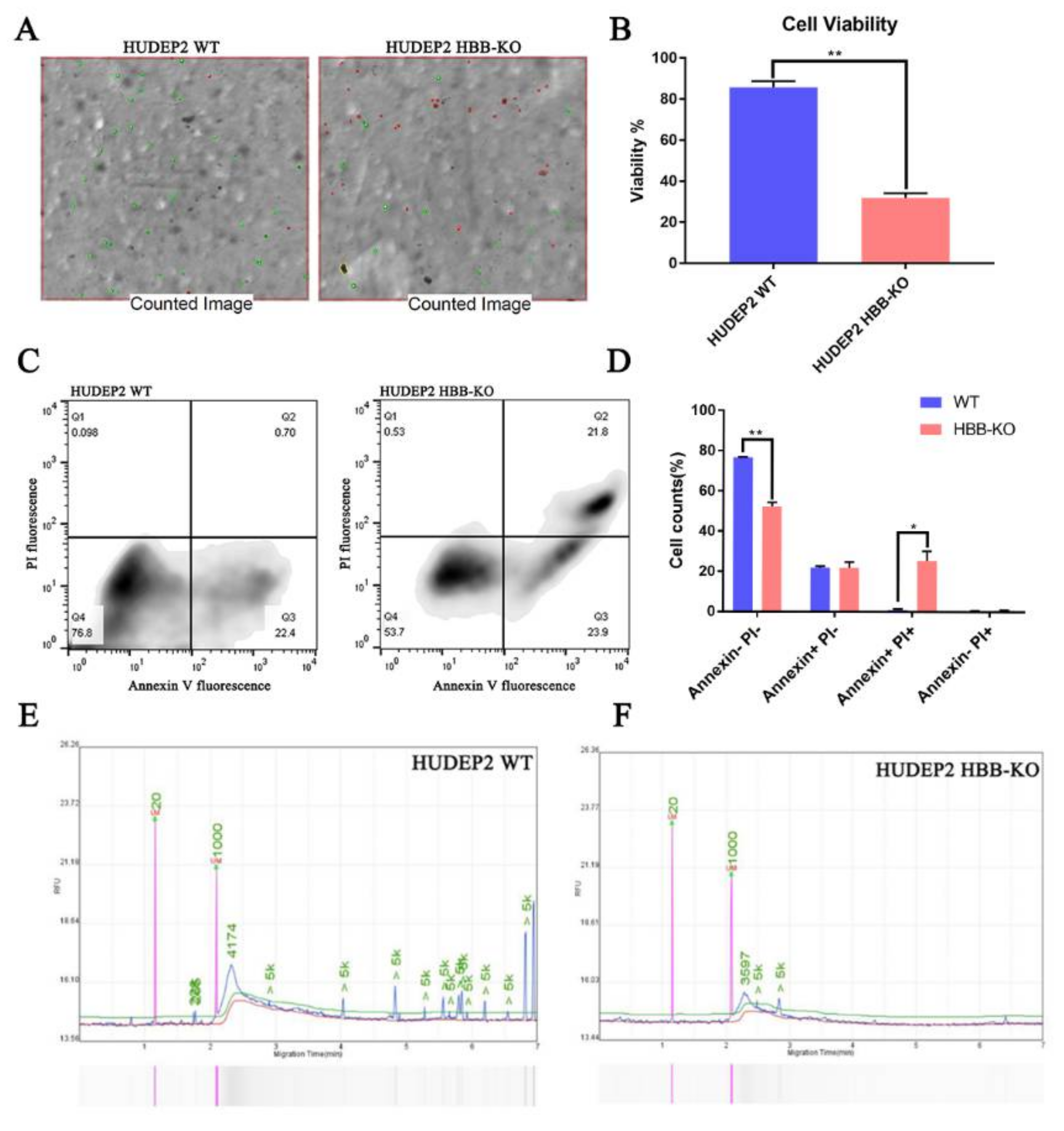
2.3. Transcriptome Analysis Indicates That Oxidative Stress May Have Occurred in HBB-KO Cells
2.4. Proteome Analysis Reveals That the HIF-1 Pathway Is Activated in HBB-KO Cells
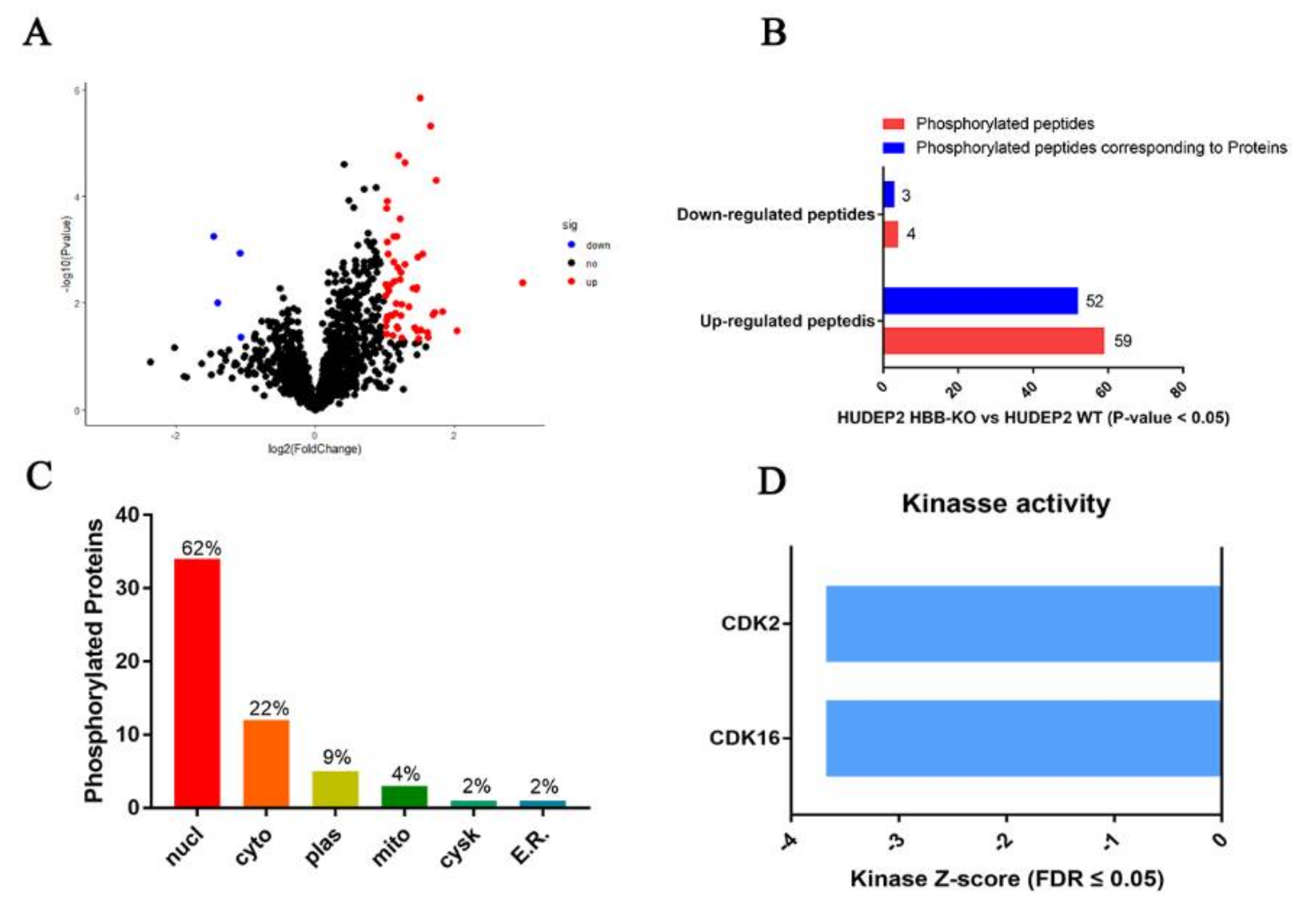
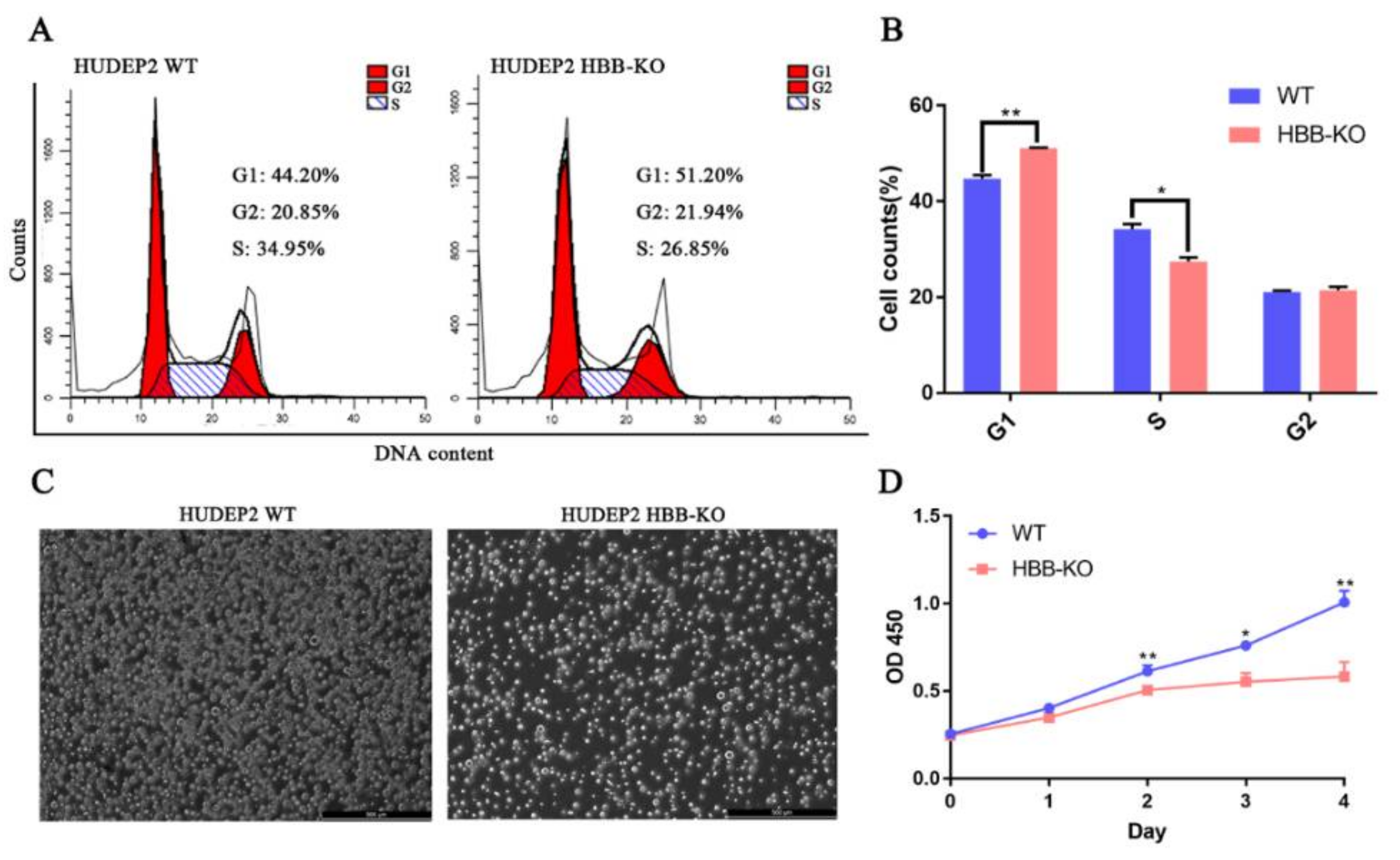
2.5. Phosphoproteome Analysis Indicates That Cell Cycle Progression May Be Slower in HBB-KO Cells
2.6. Stress Erythropoiesis Increases HBG1/2 Expression in Response to Loss of HBB
3. Discussion
4. Materials and Methods
4.1. Site-Specific Mutagenesis
4.2. RT-qPCR Assays
4.3. Cell Culture and Erythroid Differentiation
4.4. RNA-Seq
4.5. CUT&Tag and ATAC-Seq
4.6. Proteome and Phosphoproteome Analysis
4.7. Cytobiology
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kruse, A.; Uehlinger, D.E.; Gotch, F.; Kotanko, P.; Levin, N.W. Red blood cell lifespan, erythropoiesis and hemoglobin control. Hemodial.-Basic Res. Clin. Trials 2008, 161, 247–254. [Google Scholar]
- Wienert, B.; Martyn, G.E.; Funnell, A.P.; Quinlan, K.G.; Crossley, M. Wake-up sleepy gene: Reactivating fetal globin for β-hemoglobinopathies. Trends Genet. 2018, 34, 927–940. [Google Scholar] [CrossRef]
- Venkatesan, V.; Srinivasan, S.; Babu, P.; Thangavel, S. Manipulation of developmental gamma-globin gene expression: An approach for healing hemoglobinopathies. Mol. Cell. Biol. 2020, 41, e00220–e00253. [Google Scholar] [CrossRef]
- Boontanrart, M.Y.; Schröder, M.S.; Stehli, G.M.; Banović, M.; Wyman, S.K.; Lew, R.J.; Bordi, M.; Gowen, B.G.; DeWitt, M.A.; Corn, J.E. ATF4 Regulates MYB to Increase γ-Globin in Response to Loss of β-Globin. Cell Rep. 2020, 32, 107993. [Google Scholar] [CrossRef]
- Rund, D.; Rachmilewitz, E. β-Thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R.; Flavell, R. Physical mapping of the globin gene deletion in hereditary persistence of foetal haemoglobin (HPFH). Nucleic Acids Res. 1980, 8, 1521–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, J.; Craig, J.; Thein, S. Fetal hemoglobin levels in adults. Blood Rev. 1994, 8, 213–224. [Google Scholar] [CrossRef]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; Van Rooijen, N. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, R.F.; Shi, L.; Wu, D.-C. Stress erythropoiesis: New signals and new stress progenitor cells. Curr. Opin. Hematol. 2011, 18, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, J.; Wu, D.-C.; Chen, Y.; Paulson, R.F. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood J. Am. Soc. Hematol. 2015, 125, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- Alter, B. Fetal erythropoiesis in stress hematopoiesis. Exp. Hematol. 1979, 7, 200–209. [Google Scholar] [PubMed]
- Zhang, F.L.; Shen, G.M.; Liu, X.L.; Wang, F.; Zhao, Y.Z.; Zhang, J.W. Hypoxia-inducible factor 1–mediated human GATA1 induction promotes erythroid differentiation under hypoxic conditions. J. Cell. Mol. Med. 2012, 16, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 2009, 24, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.K.; Winocour, P.; Farrington, K. Erythropoietic stress and anemia in diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 204–210. [Google Scholar] [CrossRef]
- Manca, L.; Masala, B. Disorders of the synthesis of human fetal hemoglobin. IUBMB Life 2008, 60, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ding, C.; Liang, P.; Li, D.; Tang, Y.; Meng, W.; Sun, H.; Lu, H.; Chen, Y.; Chen, X. HBB-deficient Macaca fascicularis monkey presents with human β-thalassemia. Protein Cell 2019, 10, 538–542. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, M.A.; Magis, W.; Bray, N.L.; Wang, T.; Berman, J.R.; Urbinati, F.; Heo, S.-J.; Mitros, T.; Muñoz, D.P.; Boffelli, D. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016, 8, 360ra134. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Lee, C.M.; Dever, D.P.; Davis, T.H.; Camarena, J.; Srifa, W.; Zhang, Y.; Paikari, A.; Chang, A.K.; Porteus, M.H. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019, 47, 7955–7972. [Google Scholar] [CrossRef]
- Ji, P.; Murata-Hori, M.; Lodish, H.F. Formation of mammalian erythrocytes: Chromatin condensation and enucleation. Trends Cell Biol. 2011, 21, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, S.K.; Ulirsch, J.C.; Sankaran, V.G. Advances in understanding erythropoiesis: Evolving perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Werner, T.; Becher, I.; Sweetman, G.; Doce, C.; Savitski, M.M.; Bantscheff, M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. [Google Scholar] [CrossRef] [PubMed]
- Hogrebe, A.; von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.T.; Kelstrup, C.D.; Olsen, J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Bauer, D.E.; Kerenyi, M.A.; Vo, T.D.; Hou, S.; Hsu, Y.-J.; Yao, H.; Trowbridge, J.J.; Mandel, G.; Orkin, S.H. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc. Natl. Acad. Sci. USA 2013, 110, 6518–6523. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Khandros, E.; Huang, P.; Peslak, S.A.; Bhardwaj, S.K.; Grevet, J.D.; Abdulmalik, O.; Wang, H.; Keller, C.A.; Giardine, B. The E3 ligase adaptor molecule SPOP regulates fetal hemoglobin levels in adult erythroid cells. Blood Adv. 2019, 3, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Chen, T.; Ijaz, H.; Cho, E.H.; Steinberg, M.H. SIRT1 activates the expression of fetal hemoglobin genes. Am. J. Hematol. 2017, 92, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Stemmer, M.; Thumberger, T.; del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [Green Version]
- Buglo, E.; Sarmiento, E.; Martuscelli, N.B.; Sant, D.W.; Danzi, M.C.; Abrams, A.J.; Dallman, J.E.; Züchner, S. Genetic compensation in a stable slc25a46 mutant zebrafish: A case for using F0 CRISPR mutagenesis to study phenotypes caused by inherited disease. PLoS ONE 2020, 15, e0230566. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhu, P.; Shi, H.; Guo, L.; Zhang, Q.; Chen, Y.; Chen, S.; Zhang, Z.; Peng, J.; Chen, J. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 2019, 568, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Wiredja, D.D.; Koyutürk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef] [PubMed]
- Hua, P.; Roy, N.; de la Fuente, J.; Wang, G.; Thongjuea, S.; Clark, K.; Roy, A.; Psaila, B.; Ashley, N.; Harrington, Y. Single-cell analysis of bone marrow–derived CD34+ cells from children with sickle cell disease and thalassemia. Blood 2019, 134, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Kurita, R.; Suda, N.; Sudo, K.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 2013, 8, e59890. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, G. A simple process of RNA-sequence analyses by Hisat2, Htseq and DESeq2. In Proceedings of the 2017 International Conference on Biomedical Engineering and Bioinformatics, Bangkok, Thailand, 14–16 September 2017; pp. 11–15. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Van Verk, M.C.; Hickman, R.; Pieterse, C.M.; Van Wees, S.C. RNA-Seq: Revelation of the messengers. Trends Plant Sci. 2013, 18, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Factoextra: Extract and visualize the results of multivariate data analyses. R Package 2017, 76, 337–354. [Google Scholar]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package 2015, 1, 790. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, T.E.; Jørgensen, T.J.; Jensen, O.N.; Larsen, M.R. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat. Protoc. 2006, 1, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, X.; Jia, X.; Hou, W.; Zhou, G.; Ma, Z.; Yu, B.; Pi, Y.; Zhang, X.; Wang, J. Phosphorylation of Msx1 promotes cell proliferation through the Fgf9/18-MAPK signaling pathway during embryonic limb development. Nucleic Acids Res. 2020, 48, 11452–11467. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, G.; Zhang, H.; Lin, A.; Wu, Z.; Li, T.; Zhang, X.; Chen, H.; Lu, D. Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model. Int. J. Mol. Sci. 2022, 23, 2807. https://doi.org/10.3390/ijms23052807
Zhou G, Zhang H, Lin A, Wu Z, Li T, Zhang X, Chen H, Lu D. Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model. International Journal of Molecular Sciences. 2022; 23(5):2807. https://doi.org/10.3390/ijms23052807
Chicago/Turabian StyleZhou, Guoqiang, Haokun Zhang, Anning Lin, Zhen Wu, Ting Li, Xumin Zhang, Hongyan Chen, and Daru Lu. 2022. "Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model" International Journal of Molecular Sciences 23, no. 5: 2807. https://doi.org/10.3390/ijms23052807