
Non-Canonical Splicing and Its Implications in Brain Physiology and Cancer


Abstract
:1. Introduction
2. Mechanisms of Canonical Splicing: Splicing and Alternative Splicing
2.1. Molecular Mechanisms Underlying Splicing Catalysis
2.2. Alternative Splicing
2.3. Regulation of Alternative Splicing
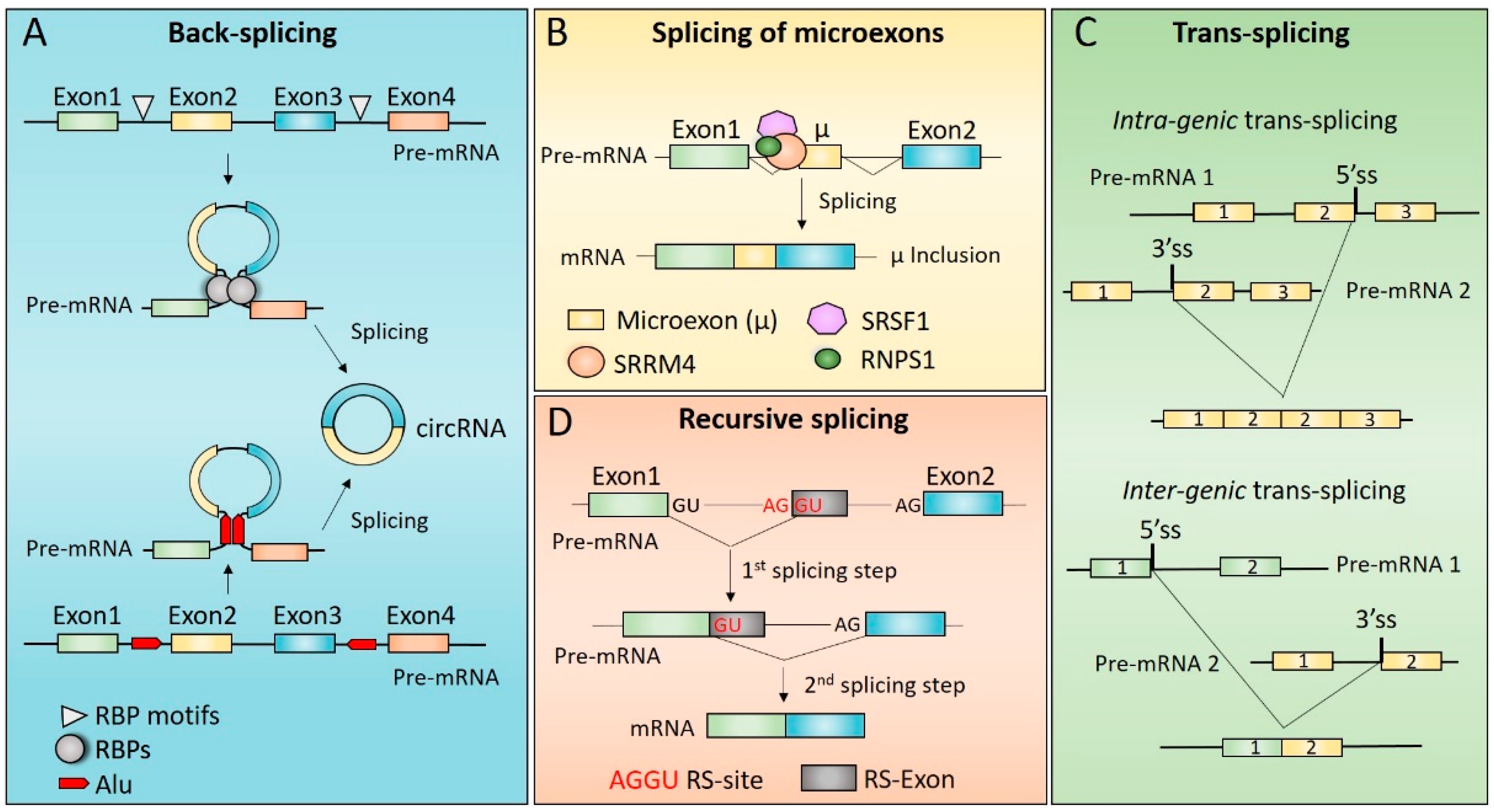
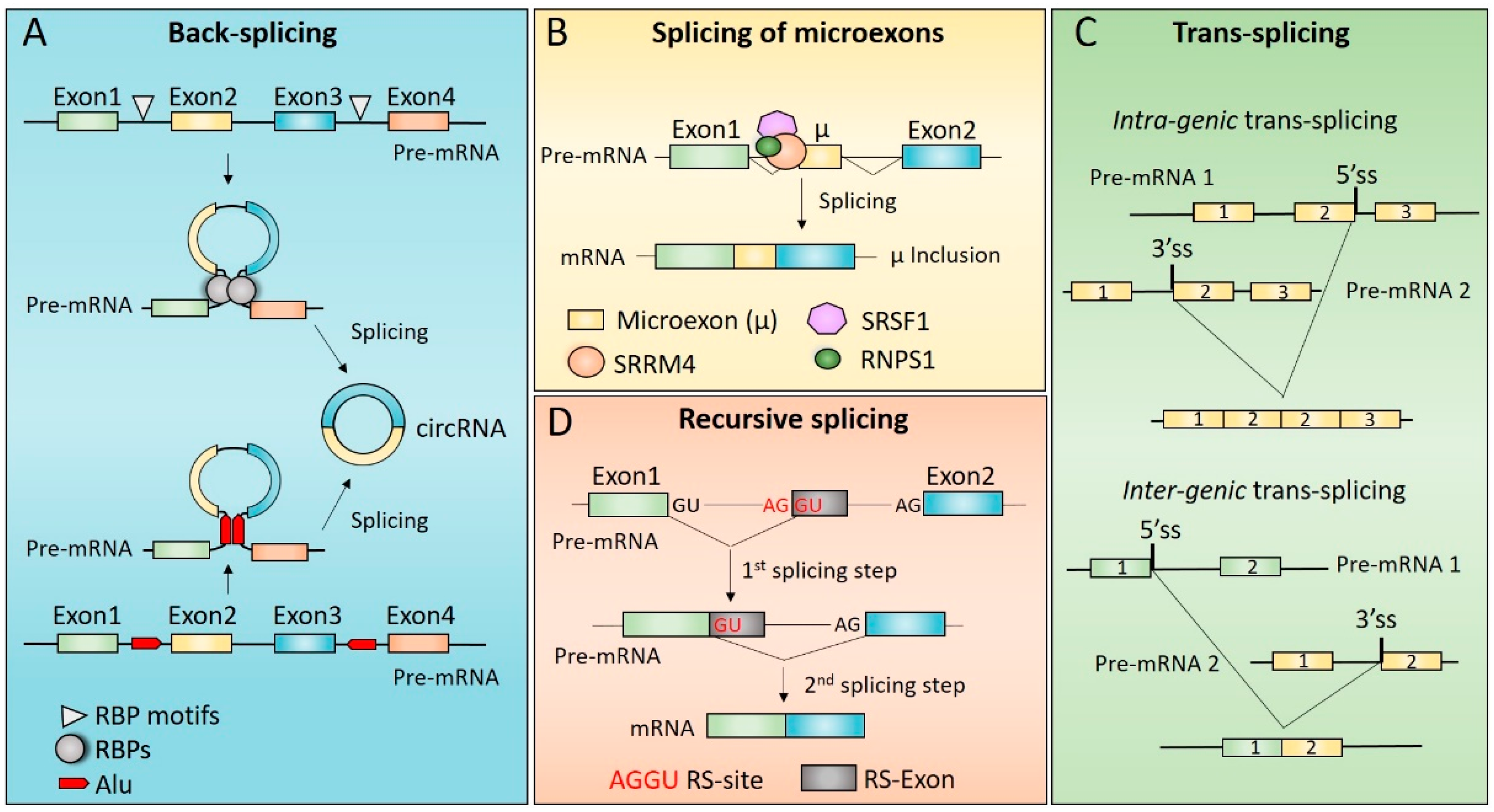
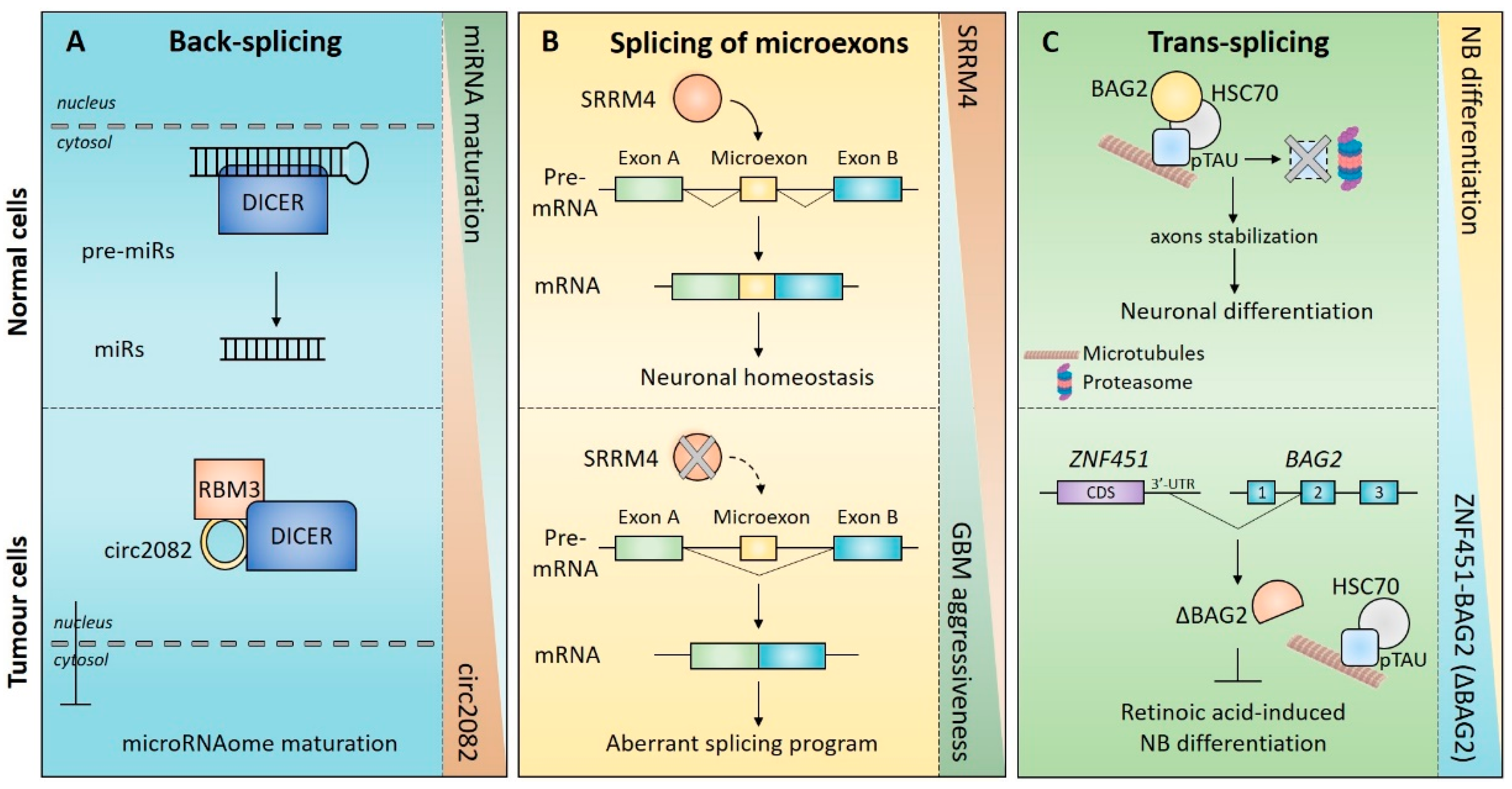
3. Mechanisms of Non-Canonical Splicing
3.1. Back-Splicing and Alternative Back-Splicing
3.1.1. Molecular Mechanisms Underlying circRNAs Biogenesis
3.1.2. Cis-Acting Elements and Trans-Acting Factors Involved in circRNAs Biogenesis
3.1.3. The Complex Crosstalk between Canonical Splicing and Back-Splicing
3.1.4. Cellular Functions of circRNAs
3.2. Splicing of Microexons
3.3. Trans-Splicing
3.3.1. Molecular Mechanisms Underlying Chimeric RNAs Biogenesis
3.3.2. Examples of Chimeric RNAs in Humans
3.4. Recursive Splicing
4. Impact of Non-Canonical Splicing in Brain Physiology
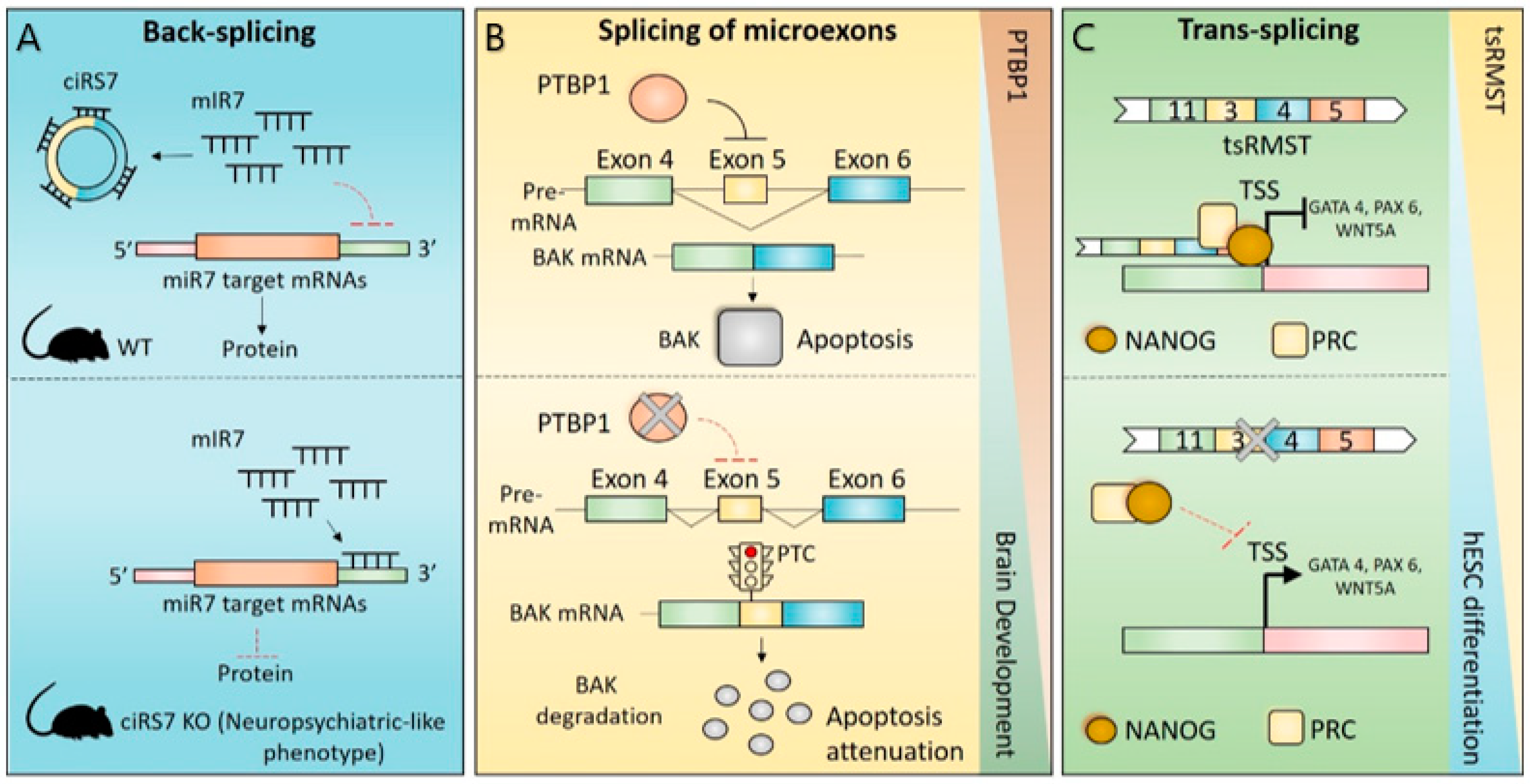
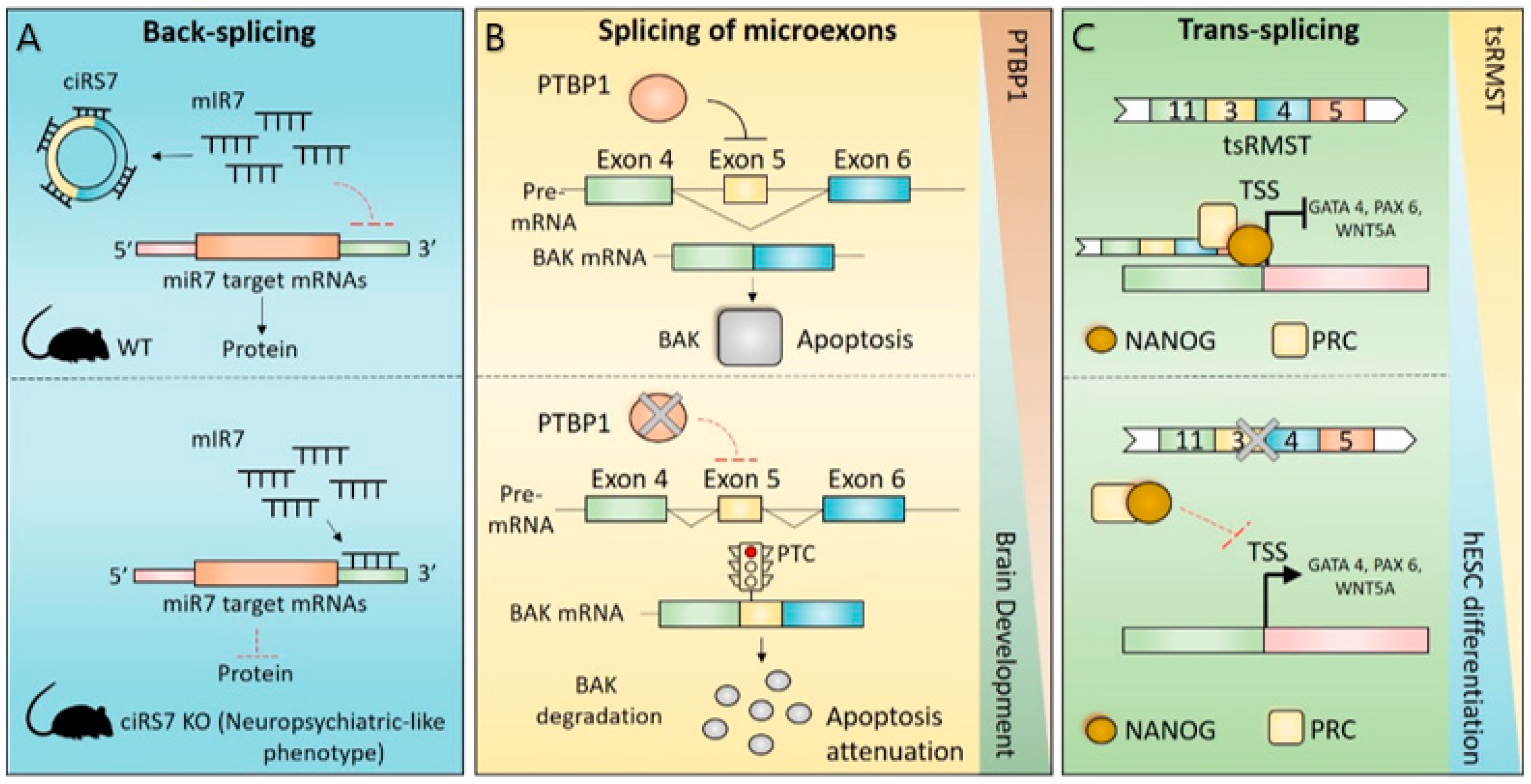
4.1. CircRNAs in Brain Physiology
4.2. Splicing of Microexons in Brain Physiology
4.3. Trans-Splicing in Brain Physiology
4.4. Recursive Splicing in Brain Physiology
5. Impact of Non-Canonical Splicing in Brain Tumours
5.1. circRNAs in Brain Tumours
5.1.1. CircRNAs in Gliomas
5.1.2. CircRNAs in Medulloblastoma
5.2. Splicing of Microexons in Brain Tumors
5.3. Trans-Splicing in Brain Tumors
6. Therapeutic Applications of Non-Canonical Splicing
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of Alternative Splicing through Coupling with Transcription and Chromatin Structure. Annu. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from Non-Canonical Splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A Highly Conserved Program of Neuronal Microexons Is Misregulated in Autistic Brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Sibley, C.R.; Emmett, W.; Blazquez, L.; Faro, A.; Haberman, N.; Briese, M.; Trabzuni, D.; Ryten, M.; Weale, M.E.; Hardy, J.; et al. Recursive Splicing in Long Vertebrate Genes. Nature 2015, 521, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duff, M.O.; Olson, S.; Wei, X.; Garrett, S.C.; Osman, A.; Bolisetty, M.; Plocik, A.; Celniker, S.E.; Graveley, B.R. Genome-Wide Identification of Zero Nucleotide Recursive Splicing in Drosophila. Nature 2015, 521, 376–379. [Google Scholar] [CrossRef] [Green Version]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Jara, C.A.C.; Fenske, P.; et al. Loss of a Mammalian Circular RNA Locus Causes MiRNA Deregulation and Affects Brain Function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef] [Green Version]
- Craig Venter, J.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Mechanistic Insights into Precursor Messenger RNA Splicing by the Spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Sheth, N.; Roca, X.; Hastings, M.L.; Roeder, T.; Krainer, A.R.; Sachidanandam, R. Comprehensive Splice-Site Analysis Using Comparative Genomics. Nucleic Acids Res. 2006, 34, 3955–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastner, B.; Will, C.L.; Stark, H.; Lührmann, R. Structural Insights into Nuclear Pre-MRNA Splicing in Higher Eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032417. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Perumal, B.S.; Sakharkar, K.R.; Kangueane, P. An Analysis on Gene Architecture in Human and Mouse Genomes. Silico Biol. 2005, 5, 347–365. [Google Scholar]
- de Conti, L.; Baralle, M.; Buratti, E. Exon and Intron Definition in Pre-MRNA Splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
- Dominski, Z.; Kole, R. Selection of Splice Sites in Pre-MRNAs with Short Internal Exons. Mol. Cell. Biol. 1991, 11, 6075–6083. [Google Scholar]
- Bentley, D.L. Coupling MRNA Processing with Transcription in Time and Space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Ameur, A.; Zaghlool, A.; Halvardson, J.; Wetterbom, A.; Gyllensten, U.; Cavelier, L.; Feuk, L. Total RNA Sequencing Reveals Nascent Transcription and Widespread Co-Transcriptional Splicing in the Human Brain. Nat. Struct. Mol. Biol. 2011, 18, 1435–1440. [Google Scholar] [CrossRef]
- Tilgner, H.; Knowles, D.G.; Johnson, R.; Davis, C.A.; Chakrabortty, S.; Djebali, S.; Curado, J.; Snyder, M.; Gingeras, T.R.; Guigó, R. Deep Sequencing of Subcellular RNA Fractions Shows Splicing to Be Predominantly Co-Transcriptional in the Human Genome but Inefficient for LncRNAs. Genome Res. 2012, 22, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- de la Mata, M.; Muñoz, M.J.; Alló, M.; Fededa, J.P.; Schor, I.E.; Kornblihtt, A.R. RNA Polymerase II Elongation at the Crossroads of Transcription and Alternative Splicing. Genet. Res. Int. 2011, 2011, 309865. [Google Scholar] [CrossRef] [Green Version]
- Corden, J.L.; Cadena, D.L.; Ahearn, J.M.; Dahmus, M.E. A Unique Structure at the Carboxyl Terminus of the Largest Subunit of Eukaryotic RNA Polymerase II. Proc. Natl. Acad. Sci. USA 1985, 82, 7934–7938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaborowska, J.; Egloff, S.; Murphy, S. The Pol II CTD: New Twists in the Tail. Nat. Struct. Mol. Biol. 2016, 23, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Eick, D.; Bensaude, O. CTD Serine-2 Plays a Critical Role in Splicing and Termination Factor Recruitment to RNA Polymerase II in Vivo. Nucleic Acids Res. 2013, 41, 1591–1603. [Google Scholar] [CrossRef] [Green Version]
- de La Mata, M.; Alonso, C.R.; Kadener, S.; Fededa, J.P.; Blaustein, M.; Pelisch, F.; Cramer, P.; Bentley, D.; Kornblihtt, A.R. A Slow RNA Polymerase II Affects Alternative Splicing In Vivo. Mol. Cell 2003, 12, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Aslanzadeh, V.; Huang, Y.; Sanguinetti, G.; Beggs, J.D. Transcription Rate Strongly Affects Splicing Fidelity and Cotranscriptionality in Budding Yeast. Genome Res. 2018, 28, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of Alternative Splicing by Histone Modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-Promoted RNA Polymerase II Pausing Links DNA Methylation to Splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf P52 Binds Methylated Histone H3K36 and Splicing Factors and Contributes to the Regulation of Alternative Splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-L. The Expanding Regulatory Mechanisms and Cellular Functions of Circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Memczak, S.; Papavasileiou, P.; Peters, O.; Rajewsky, N. Identification and Characterization of Circular RNAs As a New Class of Putative Biomarkers in Human Blood. PLoS ONE 2015, 10, e0141214. [Google Scholar] [CrossRef]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.-H.; Bindereif, A. Exon Circularization Requires Canonical Splice Signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary Sequence-Mediated Exon Circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Wilusz, J.E. Short Intronic Repeat Sequences Facilitate Circular RNA Production. Genes Dev. 2014, 28, 2233–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Zhang, X.O.; Jiang, T.; Cai, L.; Huang, X.; Liu, Q.; Li, D.; Lu, A.; Liu, Y.; Xue, W.; et al. Comprehensive Identification of Alternative Back-Splicing in Human Tissue Transcriptomes. Nucleic Acids Res. 2020, 48, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA Biogenesis Competes with Pre-MRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of CircRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Knupp, D.; Cooper, D.A.; Saito, Y.; Darnell, R.B.; Miura, P. NOVA2 Regulates Neural CircRNA Biogenesis. Nucleic Acids Res. 2021, 49, 6849–6862. [Google Scholar] [CrossRef]
- Sette, C.; Messina, V.; Paronetto, M.P. Sam68: A New STAR in the Male Fertility Firmament. J. Androl. 2010, 31, 66–74. [Google Scholar] [CrossRef]
- Pagliarini, V.; Jolly, A.; Bielli, P.; di Rosa, V.; de La Grange, P.; Sette, C. Sam68 Binds Alu-Rich Introns in SMN and Promotes Pre-MRNA Circularization. Nucleic Acids Res. 2020, 48, 633–645. [Google Scholar] [CrossRef]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of Intron Sequences Reveals Hallmarks of Circular RNA Biogenesis in Animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Aktaş, T.; Ilik, I.A.; Maticzka, D.; Bhardwaj, V.; Pessoa Rodrigues, C.; Mittler, G.; Manke, T.; Backofen, R.; Akhtar, A. DHX9 Suppresses RNA Processing Defects Originating from the Alu Invasion of the Human Genome. Nature 2017, 544, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, W.; Li, X.; Zhang, J.; Chen, S.; Zhang, J.L.; Yang, L.; Chen, L.L. The Biogenesis of Nascent Circular RNAs. Cell Rep. 2016, 15, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs Are Abundant, Conserved, and Associated with ALU Repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Tatomer, D.C.; Luo, Z.; Wu, H.; Yang, L.; Chen, L.L.; Cherry, S.; Wilusz, J.E. The Output of Protein-Coding Genes Shifts to Circular RNAs When the Pre-MRNA Processing Machinery Is Limiting. Mol. Cell 2017, 68, 940–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagsted, L.V.W.; O’leary, E.T.; Ebbesen, K.K.; Hansen, T.B. The RNA-Binding Protein SFPQ Preserves Long-Intron Splicing and Regulates CircRNA Biogenesis in Mammals. eLife 2021, 10, e63088. [Google Scholar] [CrossRef]
- Thomas-Jinu, S.; Gordon, P.M.; Fielding, T.; Taylor, R.; Smith, B.N.; Snowden, V.; Blanc, E.; Vance, C.; Topp, S.; Wong, C.H.; et al. Non-Nuclear Pool of Splicing Factor SFPQ Regulates Axonal Transcripts Required for Normal Motor Development. Neuron 2017, 94, 931. [Google Scholar] [CrossRef]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron Retention and Nuclear Loss of SFPQ Are Molecular Hallmarks of ALS. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Yu, C.Y.; Kuo, H.C. The Emerging Roles and Functions of Circular RNAs and Their Generation. J. Biomed. Sci. 2019, 26, 1–12. [Google Scholar] [CrossRef]
- Panda, A.C. Circular RNAs Act as MiRNA Sponges. Adv. Exp. Med. Biol. 2018, 1087, 67–79. [Google Scholar] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The Multilayered Complexity of CeRNA Crosstalk and Competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient MicroRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs Are a Large Class of Animal RNAs with Regulatory Potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA Profiling Reveals an Abundant CircHIPK3 That Regulates Cell Growth by Sponging Multiple MiRNAs. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Li, X.-X.; Xiao, L.; Chung, H.K.; Ma, X.-X.; Liu, X.; Song, J.-L.; Jin, C.Z.; Rao, J.N.; Gorospe, M.; Wang, J.-Y. Interaction between HuR and CircPABPN1 Modulates Autophagy in the Intestinal Epithelium by Altering ATG16L1 Translation. Mol. Cell. Biol. 2020, 40, e00492-19. [Google Scholar] [CrossRef]
- Tsitsipatis, D.; Grammatikakis, I.; Driscoll, R.K.; Yang, X.; Abdelmohsen, K.; Harris, S.C.; Yang, J.H.; Herman, A.B.; Chang, M.W.; Munk, R.; et al. AUF1 Ligand CircPCNX Reduces Cell Proliferation by Competing with P21 MRNA to Increase P21 Production. Nucleic Acids Res. 2021, 49, 1631–1646. [Google Scholar] [CrossRef]
- Gratacós, F.M.; Brewer, G. The Role of AUF1 in Regulated MRNA Decay. Wiley Interdiscip. Rev. RNA 2010, 1, 457–473. [Google Scholar] [CrossRef] [Green Version]
- Srikantan, S.; Gorospe, M. HuR Function in Disease. Front. Biosci. Landmark Ed. 2012, 17, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Yang, G.; Wang, X.; Liu, J.; Lu, Z.; Wang, Q.; Xu, B.; Liu, Z.; Li, J. CircBACH1 (Hsa_circ_0061395) Promotes Hepatocellular Carcinoma Growth by Regulating P27 Repression via HuR. J. Cell. Physiol. 2020, 235, 6929–6941. [Google Scholar] [CrossRef]
- Kullmann, M.; Göpfert, U.; Siewe, B.; Hengst, L. ELAV/Hu Proteins Inhibit P27 Translation via an IRES Element in the P27 5′UTR. Genes Dev. 2002, 16, 3087–3099. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Beltran, M.; Damizia, M.; Grelloni, C.; Colantoni, A.; Setti, A.; di Timoteo, G.; Dattilo, D.; Centrón-Broco, A.; Nicoletti, C.; et al. Circular RNA ZNF609/CKAP5 MRNA Interaction Regulates Microtubule Dynamics and Tumorigenicity. Mol. Cell 2022, 82, 75–89. [Google Scholar] [CrossRef]
- Li, B.; Zhu, L.; Lu, C.; Wang, C.; Wang, H.; Jin, H.; Ma, X.; Cheng, Z.; Yu, C.; Wang, S.; et al. CircNDUFB2 Inhibits Non-Small Cell Lung Cancer Progression via Destabilizing IGF2BPs and Activating Anti-Tumor Immunity. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-Intron Circular RNAs Regulate Transcription in the Nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, G.; Yan, X.; Lv, Z.; Yin, H.; Zhang, S.; Song, W.; Li, X.; Li, L.; Du, Z.; et al. A Novel FLI1 Exonic Circular RNA Promotes Metastasis in Breast Cancer by Coordinately Regulating TET1 and DNMT1. Genome Biol. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, J.; Sonenberg, N. Internal Initiation of Translation of Eukaryotic MRNA Directed by a Sequence Derived from Poliovirus RNA. Nature 1988, 334, 320–325. [Google Scholar] [CrossRef]
- Legnini, I.; di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA That Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fan, X.; Mao, M.; Song, X.; Wu, P.; Zhang, Y.; Jin, Y.; Yang, Y.; Chen, L.L.; Wang, Y.; et al. Extensive Translation of Circular RNAs Driven by N 6-Methyladenosine. Cell Res. 2017, 27, 626–641. [Google Scholar] [CrossRef] [Green Version]
- di Timoteo, G.; Dattilo, D.; Centrón-Broco, A.; Colantoni, A.; Guarnacci, M.; Rossi, F.; Incarnato, D.; Oliviero, S.; Fatica, A.; Morlando, M.; et al. Modulation of CircRNA Metabolism by m 6 A Modification. Cell Rep. 2020, 31, 107641. [Google Scholar] [CrossRef]
- Begum, S.; Yiu, A.; Stebbing, J.; Castellano, L. Novel Tumour Suppressive Protein Encoded by Circular RNA, circ-SHPRH, in Glioblastomas. Oncogene. 2018, 37, 4055–4057. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A Novel Protein Encoded by the Circular Form of the SHPRH Gene Suppresses Glioma Tumorigenesis. Oncogene 2018, 37, 1805–1814. [Google Scholar] [CrossRef]
- Ho-Xuan, H.; GlaÅar, P.; Latini, C.; Heizler, K.; Haase, J.; Hett, R.; Anders, M.; Weichmann, F.; Bruckmann, A.; van den Berg, D.; et al. Comprehensive Analysis of Translation from Overexpressed Circular RNAs Reveals Pervasive Translation from Linear Transcripts. Nucleic Acids Res. 2020, 48, 10368–10382. [Google Scholar] [CrossRef]
- Ustianenko, D.; Weyn-Vanhentenryck, S.M.; Zhang, C. Microexons: Discovery, Regulation, and Function. Wiley Interdiscip. Rev. RNA 2017, 8, e1418. [Google Scholar] [CrossRef]
- Gonatopoulos-Pournatzis, T.; Blencowe, B.J. Microexons: At the Nexus of Nervous System Development, Behaviour and Autism Spectrum Disorder. Curr. Opin. Genet. Dev. 2020, 65, 22–33. [Google Scholar] [CrossRef]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 Proteins Regulate the Alternative Splicing of Micro-Exons in Human Brain Transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Parras, A.; Anta, H.; Santos-Galindo, M.; Swarup, V.; Elorza, A.; Nieto-González, J.L.; Picó, S.; Hernández, I.H.; Díaz-Hernández, J.I.; Belloc, E.; et al. Autism-like Phenotype and Risk Gene MRNA Deadenylation by CPEB4 Mis-Splicing. Nature 2018, 560, 441–446. [Google Scholar] [CrossRef]
- Parada, G.E.; Munita, R.; Georgakopoulos-Soares, I.; Fernandes, H.J.R.; Kedlian, V.R.; Metzakopian, E.; Andres, M.E.; Miska, E.A.; Hemberg, M. MicroExonator Enables Systematic Discovery and Quantification of Microexons across Mouse Embryonic Development. Genome Biol. 2021, 22, 1–26. [Google Scholar] [CrossRef]
- Raj, B.; Irimia, M.; Braunschweig, U.; Sterne-Weiler, T.; O’Hanlon, D.; Lin, Z.Y.; Chen, G.I.; Easton, L.E.; Ule, J.; Gingras, A.C.; et al. A Global Regulatory Mechanism for Activating an Exon Network Required for Neurogenesis. Mol. Cell 2014, 56, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Gonatopoulos-Pournatzis, T.; Wu, M.; Braunschweig, U.; Roth, J.; Han, H.; Best, A.J.; Raj, B.; Aregger, M.; O’Hanlon, D.; Ellis, J.D.; et al. Genome-Wide CRISPR-Cas9 Interrogation of Splicing Networks Reveals a Mechanism for Recognition of Autism-Misregulated Neuronal Microexons. Mol. Cell 2018, 72, 510–524. [Google Scholar] [CrossRef] [Green Version]
- Torres-Méndez, A.; Bonnal, S.; Marquez, Y.; Roth, J.; Iglesias, M.; Permanyer, J.; Almudí, I.; O’Hanlon, D.; Guitart, T.; Soller, M.; et al. A Novel Protein Domain in an Ancestral Splicing Factor Drove the Evolution of Neural Microexons. Nat. Ecol. Evol. 2019, 3, 691–701. [Google Scholar] [CrossRef]
- Blazquez, L.; Emmett, W.; Faraway, R.; Pineda, J.M.B.; Bajew, S.; Gohr, A.; Haberman, N.; Sibley, C.R.; Bradley, R.K.; Irimia, M.; et al. Exon Junction Complex Shapes the Transcriptome by Repressing Recursive Splicing. Mol. Cell 2018, 72, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Boehm, V.; Britto-Borges, T.; Steckelberg, A.L.; Singh, K.K.; Gerbracht, J.V.; Gueney, E.; Blazquez, L.; Altmüller, J.; Dieterich, C.; Gehring, N.H. Exon Junction Complexes Suppress Spurious Splice Sites to Safeguard Transcriptome Integrity. Mol. Cell 2018, 72, 482–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agabian, N. Trans Splicing of Nuclear Pre-MRNAs. Cell 1990, 61, 1157–1160. [Google Scholar] [CrossRef]
- Wang, Y.; Zou, Q.; Li, F.; Zhao, W.; Xu, H.; Zhang, W.; Deng, H.; Yang, X. Identification of the Cross-Strand Chimeric RNAs Generated by Fusions of Bi-Directional Transcripts. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Rabbitts, T.H. Chromosomal Translocations in Human Cancer. Nature 1994, 372, 143–149. [Google Scholar] [CrossRef]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA Trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Xie, Y.; Martignetti, J.A.; Yeo, T.T.; Massa, S.M.; Longo, F.M. A Candidate Chimeric Mammalian MRNA Transcript Is Derived from Distinct Chromosomes and Is Associated with Nonconsensus Splice Junction Motifs. DNA Cell Biol. 2003, 22, 303–315. [Google Scholar] [CrossRef]
- Li, X.; Zhao, L.; Jiang, H.; Wang, W. Short Homologous Sequences Are Strongly Associated with the Generation of Chimeric RNAs in Eukaryotes. J. Mol. Evol. 2009, 68, 56–65. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, Y.; Li, X.; Jia, Y.; Li, F.; Li, J.; Zhang, Z. Evidence of Constraint in the 3D Genome for Trans-Splicing in Human Cells. Sci. China Life Sci. 2020, 63, 1380–1393. [Google Scholar] [CrossRef]
- Williams, A.; Flavell, R.A. The Role of CTCF in Regulating Nuclear Organization. J. Exp. Med. 2008, 205, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Yang, S.; Zhao, W.; Tang, Z.; Zhang, T.; Li, K. Identification and Analysis of Pig Chimeric MRNAs Using RNA Sequencing Data. BMC Genom. 2012, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, D.; Trajanoski, Z.; McNally, J.G. Transcription Factories. Front. Genet. 2012, 3, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitar, M.; Boroni, M.; Macedo, A.M.; Machado, C.R.; Franco, G.R. The Spliced Leader Trans-Splicing Mechanism in Different Organisms: Molecular Details and Possible Biological Roles. Front. Genet. 2013, 4, 199. [Google Scholar] [CrossRef] [Green Version]
- Herai, R.H.; Yamagishi, M.E.B. Detection of Human Interchromosomal Trans-Splicing in Sequence Databanks. Brief. Bioinform. 2010, 11, 198–209. [Google Scholar] [CrossRef]
- Frenkel-Morgenstern, M.; Gorohovski, A.; Lacroix, V.; Rogers, M.; Ibanez, K.; Boullosa, C.; Leon, E.A.; Ben-Hur, A.; Valencia, A. ChiTaRS: A Database of Human, Mouse and Fruit Fly Chimeric Transcripts and RNA-Sequencing Data. Nucleic Acids Res. 2013, 41, D142–D151. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Ma, X.; Sklar, J. Gene Fusions and RNA Trans-Splicing in Normal and Neoplastic Human Cells. Cell Cycle 2009, 8, 218–222. [Google Scholar] [CrossRef]
- Li, N.; Zheng, J.; Li, H.; Deng, J.; Hu, M.; Wu, H.; Li, W.; Li, F.; Lan, X.; Lu, J.; et al. Identification of Chimeric TSNAX-DISC1 Resulting from Intergenic Splicing in Endometrial Carcinoma through High-Throughput RNA Sequencing. Carcinogenesis 2014, 35, 2687–2697. [Google Scholar] [CrossRef] [Green Version]
- Koontz, J.I.; Soreng, A.L.; Nucci, M.; Kuo, F.C.; Pauwels, P.; van den Berghe, H.; Dal Cin, P.; Fletcher, J.A.; Sklar, J. Frequent Fusion of the JAZF1 and JJAZ1 Genes in Endometrial Stromal Tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 6348–6353. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A Neoplastic Gene Fusion Mimics Trans-Splicing of RNAs in Normal Human Cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef]
- Nucci, M.R.; Harburger, D.; Koontz, J.; Cin, P.D.; Sklar, J. Molecular Analysis of the JAZF1-JJAZ1 Gene Fusion by RT-PCR and Fluorescence in Situ Hybridization in Endometrial Stromal Neoplasms. Am. J. Surg. Pathol. 2007, 31, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xian, Y.M.; Wang, J.; Koontz, J.; Nucci, M.; Sklar, J. Effects of Rearrangement and Allelic Exclusion of JJAZ1/SUZ12 on Cell Proliferation and Survival. Proc. Natl. Acad. Sci. USA 2007, 104, 20001–20006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Babiceanu, M.; Kumar, S.; Jia, Y.; Qin, F.; Barr, F.G.; Li, H. Fusion Transcriptome Profiling Provides Insights Into Alveolar Rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA. 2016, 113, 13126–13131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A Chimeric RNA Characteristic of Rhabdomyosarcoma in Normal Myogenesis Process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linardic, C.M. PAX3–FOXO1 Fusion Gene in Rhabdomyosarcoma. Cancer Lett. 2008, 270, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Li, X.; Li, H. Gene Fusions and Chimeric RNAs, and Their Implications in Cancer. Genes Dis. 2019, 6, 385–390. [Google Scholar] [CrossRef]
- Mochizuki, K. DNA Rearrangements Directed by Non-Coding RNAs in Ciliates. Wiley Interdiscip. Rev. RNA 2010, 1, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.K.; Luo, L.; Yen, L. RNA-Mediated Gene Fusion in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E12295–E12304. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Hatton, A.R.; Subramaniam, V.; Lopez, A.J. Generation of Alternative Ultrabithorax Isoforms and Stepwise Removal of a Large Intron by Resplicing at Exon-Exon Junctions. Mol. Cell 1998, 2, 787–796. [Google Scholar] [CrossRef]
- Burnette, J.M.; Miyamoto-Sato, E.; Schaub, M.A.; Conklin, J.; Lopez, A.J. Subdivision of Large Introns in Drosophila by Recursive Splicing at Nonexonic Elements. Genetics 2005, 170, 661–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, B.; Lai, E.C. The Exon Junction Complex and Intron Removal Prevent Re-Splicing of MRNA. PLoS Genet. 2021, 17, e1009563. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, H.; Saulière, J.; Wang, Z. The Exon Junction Complex as a Node of Post-Transcriptional Networks. Nat. Rev. Mol. Cell Biol. 2016, 17, 41–54. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural Circular RNAs Are Derived from Synaptic Genes and Regulated by Development and Plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. MiRNA-Dependent Gene Silencing Involving Ago2-Mediated Cleavage of a Circular Antisense RNA. EMBO J. 2011, 30, 4414–4422. [Google Scholar] [CrossRef] [Green Version]
- van Spronsen, M.; Hoogenraad, C.C. Synapse Pathology in Psychiatric and Neurologic Disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Hollensen, A.K.; Thomsen, H.S.; Lloret-Llinares, M.; Bjerregaard Kamstrup, A.; Malte Jensen, J.; Luckmann, M.; Birkmose, N.; Palmfeldt, J.; Jensen, T.H.; Hansen, T.B.; et al. CircZNF827 Nucleates a Transcription Inhibitory Complex to Balance Neuronal Differentiation. eLife 2020, 9, e58478. [Google Scholar] [CrossRef]
- Xu, K.; Chen, D.; Wang, Z.; Ma, J.; Zhou, J.; Chen, N.; Lv, L.; Zheng, Y.; Hu, X.; Zhang, Y.; et al. Annotation and Functional Clustering of CircRNA Expression in Rhesus Macaque Brain during Aging. Cell Discov. 2018, 4, 1–18. [Google Scholar] [CrossRef]
- Gokool, A.; Anwar, F.; Voineagu, I. The Landscape of Circular RNA Expression in the Human Brain. Biol. Psychiatry 2020, 87, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Zhang, Y.; Xiong, W.; Zhang, Z.; Wang, Z.; Lv, L.; Liu, C.; Hu, Z.; Zheng, Y.T.; Lu, L.; et al. CircGRIA1 Shows an Age-Related Increase in Male Macaque Brain and Regulates Synaptic Plasticity and Synaptogenesis. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Villarreal, O.D.; Chen, X.; Zandee, S.; Young, Y.K.; Torok, C.; Lamarche-Vane, N.; Prat, A.; Rivest, S.; Gosselin, D.; et al. QUAKING Regulates Microexon Alternative Splicing of the Rho GTPase Pathway and Controls Microglia Homeostasis. Cell Rep. 2020, 33, 108560. [Google Scholar] [CrossRef]
- Choudhary, B.; Marx, O.; Norris, A.D. Spliceosomal Component PRP-40 Is a Central Regulator of Microexon Splicing. Cell Rep. 2021, 36, 109464. [Google Scholar] [CrossRef] [PubMed]
- Quesnel-Vallières, M.; Irimia, M.; Cordes, S.P.; Blencowe, B.J. Essential Roles for the Splicing Regulator NSR100/SRRM4 during Nervous System Development. Genes Dev. 2015, 29, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Quesnel-Vallières, M.; Dargaei, Z.; Irimia, M.; Gonatopoulos-Pournatzis, T.; Ip, J.Y.; Wu, M.; Sterne-Weiler, T.; Nakagawa, S.; Woodin, M.A.; Blencowe, B.J.; et al. Misregulation of an Activity-Dependent Splicing Network as a Common Mechanism Underlying Autism Spectrum Disorders. Mol. Cell 2016, 64, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Gonatopoulos-Pournatzis, T.; Niibori, R.; Salter, E.W.; Weatheritt, R.J.; Tsang, B.; Farhangmehr, S.; Liang, X.; Braunschweig, U.; Roth, J.; Zhang, S.; et al. Autism-Misregulated EIF4G Microexons Control Synaptic Translation and Higher Order Cognitive Functions. Mol. Cell 2020, 77, 1176–1192. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Südhof, T.C. A Transcriptionally [Correction of Transcriptively] Active Complex of APP with Fe65 and Histone Acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Hu, Q.; Hearn, M.G.; Shimizu, K.; Ware, C.B.; Liggitt, D.H.; Jin, L.W.; Cool, B.H.; Storm, D.R.; Martin, G.M. Isoform-Specific Knockout of FE65 Leads to Impaired Learning and Memory. J. Neurosci. Res. 2004, 75, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Tsyba, L.; Gryaznova, T.; Dergai, O.; Dergai, M.; Skrypkina, I.; Kropyvko, S.; Boldyryev, O.; Nikolaienko, O.; Novokhatska, O.; Rynditch, A. Alternative Splicing Affecting the SH3A Domain Controls the Binding Properties of Intersectin 1 in Neurons. Biochem. Biophys. Res. Commun. 2008, 372, 929–934. [Google Scholar] [CrossRef]
- Saita, S.; Shirane, M.; Natume, T.; Iemura, S.I.; Nakayama, K.I. Promotion of Neurite Extension by Protrudin Requires Its Interaction with Vesicle-Associated Membrane Protein-Associated Protein. J. Biol. Chem. 2009, 284, 13766–13777. [Google Scholar] [CrossRef] [Green Version]
- Pechstein, A.; Shupliakov, O.; Haucke, V. Intersectin 1: A Versatile Actor in the Synaptic Vesicle Cycle. Biochem. Soc. Trans. 2010, 38, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Zhang, M.; Stoilov, P.; Chen, L.; Zheng, S. Developmental Attenuation of Neuronal Apoptosis by Neural-Specific Splicing of Bak1 Microexon. Neuron 2020, 107, 1180–1196. [Google Scholar] [CrossRef]
- Rusconi, F.; Grillo, B.; Toffolo, E.; Mattevi, A.; Battaglioli, E. NeuroLSD1: Splicing-Generated Epigenetic Enhancer of Neuroplasticity. Trends Neurosci. 2017, 40, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Shirane, M.; Nakayama, K.I. SRRM4-Dependent Neuron-Specific Alternative Splicing of Protrudin Transcripts Regulates Neurite Outgrowth. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, T.; Shirane, M.; Hashimoto, Y.; Saita, S.; Nakayama, K.I. Identification and Characterization of a Neuron-Specific Isoform of Protrudin. Genes Cells 2014, 19, 97–111. [Google Scholar] [CrossRef]
- Capponi, S.; Stöffler, N.; Irimia, M.; van Schaik, F.M.A.; Ondik, M.M.; Biniossek, M.L.; Lehmann, L.; Mitschke, J.; Vermunt, M.W.; Creyghton, M.P.; et al. Neuronal-Specific Microexon Splicing of TAF1 MRNA Is Directly Regulated by SRRM4/NSR100. RNA Biol. 2020, 17, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Telese, F.; Tan, Y.; Li, W.; Jin, C.; He, X.; Basnet, H.; Ma, Q.; Merkurjev, D.; Zhu, X.; et al. LSD1n Is an H4K20 Demethylase Regulating Memory Formation via Transcriptional Elongation Control. Nat. Neurosci. 2015, 18, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Chwalenia, K.; Facemire, L.; Li, H. Chimeric RNAs in Cancer and Normal Physiology. Wiley Interdiscip. Rev. RNA 2017, 8, e1427. [Google Scholar] [CrossRef]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent Chimeric Fusion RNAs in Non-Cancer Tissues and Cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Qin, F.; Kumar, S.; Elfman, J.; Lin, E.; Pham, L.P.; Yang, A.; Li, H. The Landscape of Chimeric RNAs in Non-Diseased Tissues and Cells. Nucleic Acids Res. 2020, 48, 1764–1778. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.S.; Yu, C.Y.; Chuang, C.Y.; Hsiao, M.; Kao, C.F.; Kuo, H.C.; Chuang, T.J. Integrative Transcriptome Sequencing Identifies Trans-Splicing Events with Important Roles in Human Embryonic Stem Cell Pluripotency. Genome Res. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.Y.; Kuo, H.C. The Trans-Spliced Long Noncoding RNA TsRMST Impedes Human Embryonic Stem Cell Differentiation Through WNT5A-Mediated Inhibition of the Epithelial-to-Mesenchymal Transition. Stem Cells 2016, 34, 2052–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhde, C.W.; Vives, J.; Jaeger, I.; Li, M. Rmst Is a Novel Marker for the Mouse Ventral Mesencephalic Floor Plate and the Anterior Dorsal Midline Cells. PLoS ONE 2010, 5, e8641. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The Long Noncoding RNA RMST Interacts with SOX2 to Regulate Neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, R.; Valotta, M.; Ferri, A.L.M.; Latorre, E.; Mariani, J.; Giachino, C.; Lancini, C.; Tosetti, V.; Ottolenghi, S.; Taylor, V.; et al. Hippocampal Development and Neural Stem Cell Maintenance Require Sox2-Dependent Regulation of Shh. Nat. Neurosci. 2009, 12, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.N.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular Genetics of Human Primary Microcephaly: An Overview. BMC Med. Genom. 2015, 8, S4. [Google Scholar] [CrossRef] [Green Version]
- McMahon, J.J.; Miller, E.E.; Silver, D.L. The Exon Junction Complex in Neural Development and Neurodevelopmental Disease. Int. J. Dev. Neurosci. 2016, 55, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Filippenkov, I.B.; Sudarkina, O.Y.; Limborska, S.A.; Dergunova, L.V. Multi-Step Splicing of Sphingomyelin Synthase Linear and Circular RNAs. Gene 2018, 654, 14–22. [Google Scholar] [CrossRef]
- Wan, Y.; Anastasakis, D.G.; Rodriguez, J.; Palangat, M.; Gudla, P.; Zaki, G.; Tandon, M.; Pegoraro, G.; Chow, C.C.; Hafner, M.; et al. Dynamic Imaging of Nascent RNA Reveals General Principles of Transcription Dynamics and Stochastic Splice Site Selection. Cell 2021, 184, 2878–2895. [Google Scholar] [CrossRef]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Xiang, Y.; Ko, J.; Li, S.; Jing, Y.; Zhu, X.; Ye, Y.; Zhang, Z.; Mills, T.; Feng, J.; et al. Comprehensive Characterization of Circular RNAs in ~1000 Human Cancer Cell Lines. Genome Med. 2019, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in Cancer: Opportunities and Challenges in the Field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Schuman, E. Circular RNAs in Brain and Other Tissues: A Functional Enigma. Trends Neurosci. 2016, 39, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, N.; Han, P.; Moon, B.S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA Profile in Gliomas Revealed by Identification Tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef]
- Zhu, J.; Ye, J.; Zhang, L.; Xia, L.; Hu, H.; Jiang, H.; Wan, Z.; Sheng, F.; Ma, Y.; Li, W.; et al. Differential Expression of Circular RNAs in Glioblastoma Multiforme and Its Correlation with Prognosis. Transl. Oncol. 2017, 10, 271–279. [Google Scholar] [CrossRef]
- Xiang Wang, H.; Lin Huang, Q.; Yan Shen, J.; Xu, T.; Hong, F.; Yu Gong, Z.; Li, F.; Yan, Y.; Xiang Chen, J. Expression Profile of Circular RNAs in IDH-Wild Type Glioblastoma Tissues. Clin. Neurol. Neurosurg. 2018, 171, 168–173. [Google Scholar] [CrossRef]
- Bielli, P.; Pagliarini, V.; Pieraccioli, M.; Caggiano, C.; Sette, C. Splicing Dysregulation as Oncogenic Driver and Passenger Factor in Brain Tumors. Cells 2019, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, S.; Chen, X.; Li, N.; Li, J.; Jia, R.; Pan, Y.; Liang, H. CircNT5E Acts as a Sponge of MiR-422a to Promote Glioblastoma Tumorigenesis. Cancer Res. 2018, 78, 4812–4825. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Yang, H.; Han, K.; Zhu, D.; Lun, P.; Zhao, Y. A Novel Circular RNA, Hsa_circ_0046701, Promotes Carcinogenesis by Increasing the Expression of MiR-142-3p Target ITGB8 in Glioma. Biochem. Biophys. Res. Commun. 2018, 498, 254–261. [Google Scholar] [CrossRef]
- Jin, P.; Huang, Y.; Zhu, P.; Zou, Y.; Shao, T.; Wang, O. CircRNA CircHIPK3 Serves as a Prognostic Marker to Promote Glioma Progression by Regulating MiR-654/IGF2BP3 Signaling. Biochem. Biophys. Res. Commun. 2018, 503, 1570–1574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Q.; Liao, Q. CircHIPK3: A Promising Cancer-related Circular RNA. Am. J. Transl. Res. 2020, 12, 6694–6704. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, F.; Shi, Z.; Zhao, Y.; Tian, J. CircRNA Hsa-Circ-0014359 Promotes Glioma Progression by Regulating MiR-153/PI3K Signaling. Biochem. Biophys. Res. Commun. 2019, 510, 614–620. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, Y.; Qi, L.; Ding, L.; Jiang, H.; Yu, H. NFIX Circular RNA Promotes Glioma Progression by Regulating MiR-34a-5p via Notch Signaling Pathway. Front. Mol. Neurosci. 2018, 11, 225. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Zhao, L.; Liu, Y.; Liu, X.; Zheng, J.; Yu, H.; Cai, H.; Ma, J.; Liu, L.; Wang, P.; et al. Circ-SHKBP1 Regulates the Angiogenesis of U87 Glioma-Exposed Endothelial Cells through MiR-544a/FOXP1 and MiR-379/FOXP2 Pathways. Mol. Therapy. Nucleic Acids 2018, 10, 331–348. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Zhao, L.; Liu, X.; Zheng, J.; Liu, Y.; Liu, L.; Ma, J.; Cai, H.; Li, Z.; Xue, Y. MOV10 Binding Circ-DICER1 Regulates the Angiogenesis of Glioma via MiR-103a-3p/MiR-382-5p Mediated ZIC4 Expression Change. J. Exp. Clin. Cancer Res. 2019, 38, 1–17. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Ruan, X.; Liu, X.; Zheng, J.; Liu, Y.; Liu, L.; Ma, J.; Shao, L.; Wang, D.; Shen, S.; et al. FUS/Circ_002136/MiR-138-5p/SOX13 Feedback Loop Regulates Angiogenesis in Glioma. J. Exp. Clin. Cancer Res. 2019, 38, 1–19. [Google Scholar] [CrossRef]
- Barbagallo, D.; Caponnetto, A.; Brex, D.; Mirabella, F.; Barbagallo, C.; Lauretta, G.; Morrone, A.; Certo, F.; Broggi, G.; Caltabiano, R.; et al. CircSMARCA5 Regulates VEGFA MRNA Splicing and Angiogenesis in Glioblastoma Multiforme Through the Binding of SRSF1. Cancers 2019, 11, 194. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, D.; Caponnetto, A.; Cirnigliaro, M.; Brex, D.; Barbagallo, C.; D’Angeli, F.; Morrone, A.; Caltabiano, R.; Barbagallo, G.M.; Ragusa, M.; et al. CircSMARCA5 Inhibits Migration of Glioblastoma Multiforme Cells by Regulating a Molecular Axis Involving Splicing Factors SRSF1/SRSF3/PTB. Int. J. Mol. Sci. 2018, 19, 480. [Google Scholar] [CrossRef] [Green Version]
- Bronisz, A.; Rooj, A.K.; Krawczynski, K.; Peruzzi, P.; Salinska, E.; Nakano, I.; Purow, B.; Chiocca, E.A.; Godlewski, J. The Nuclear DICER-Circular RNA Complex Drives the Deregulation of the Glioblastoma Cell MicroRNAome. Sci. Adv. 2020, 6, eabc0221. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z. Efficient Backsplicing Produces Translatable Circular MRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Gao, X.; Zhang, M.; Yan, S.; Sun, C.; Xiao, F.; Huang, N.; Yang, X.; Zhao, K.; Zhou, H.; et al. Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhao, K.; Xu, X.; Yang, Y.; Yan, S.; Wei, P.; Liu, H.; Xu, J.; Xiao, F.; Zhou, H.; et al. A Peptide Encoded by Circular Form of LINC-PINT Suppresses Oncogenic Transcriptional Elongation in Glioblastoma. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Li, X.; Li, F.; Wu, X.; Zhang, M.; Zhou, H.; Huang, N.; Yang, X.; Xiao, F.; Liu, D.; et al. A Novel Tumor Suppressor Protein Encoded by Circular AKT3 RNA Inhibits Glioblastoma Tumorigenicity by Competing with Active Phosphoinositide-Dependent Kinase-1. Mol. Cancer 2019, 18, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Xia, X.; Li, F.; Zhang, M.; Zhou, H.; Wu, X.; Zhong, J.; Zhao, Z.; Zhao, K.; Liu, D.; et al. Circular RNA-Encoded Oncogenic E-Cadherin Variant Promotes Glioblastoma Tumorigenicity through Activation of EGFR-STAT3 Signalling. Nat. Cell Biol. 2021, 23, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; De Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent Noncoding U1 SnRNA Mutations Drive Cryptic Splicing in SHH Medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Dubuc, A.M.; Morrissy, A.S.; Kloosterhof, N.K.; Northcott, P.A.; Yu, E.P.Y.; Shih, D.; Peacock, J.; Grajkowska, W.; van Meter, T.; Eberhart, C.G.; et al. Subgroup-Specific Alternative Splicing in Medulloblastoma. Acta Neuropathol. 2012, 123, 485–499. [Google Scholar] [CrossRef] [Green Version]
- Menghi, F.; Jacques, T.S.; Barenco, M.; Schwalbe, E.C.; Clifford, S.C.; Hubank, M.; Ham, J. Genome-Wide Analysis of Alternative Splicing in Medulloblastoma Identifies Splicing Patterns Characteristic of Normal Cerebellar Development. Cancer Res. 2011, 71, 2045–2055. [Google Scholar] [CrossRef] [Green Version]
- Lv, T.; Miao, Y.F.; Jin, K.; Han, S.; Xu, T.Q.; Qiu, Z.L.; Zhang, X.H. Dysregulated Circular RNAs in Medulloblastoma Regulate Proliferation and Growth of Tumor Cells via Host Genes. Cancer Med. 2018, 7, 6147–6157. [Google Scholar] [CrossRef] [PubMed]
- Azatyan, A.; Zhang, S.; Darabi, A.; Siesjö, P.; Wang, T.; Zaphiropoulos, P.G. Circular RNAs in Hedgehog Signaling Activation and Hedgehog-Mediated Medulloblastoma Tumors. Cancers 2021, 13, 5138. [Google Scholar] [CrossRef] [PubMed]
- Rickert, D.; Bartl, J.; Picard, D.; Bernardi, F.; Qin, N.; Lovino, M.; Puget, S.; Meyer, F.D.; Mahoungou Koumba, I.; Beez, T.; et al. Circular RNA Profiling Distinguishes Medulloblastoma Groups and Shows Aberrant RMST Overexpression in WNT Medulloblastoma. Acta Neuropathol. 2021, 141, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Head, S.A.; Hernandez-Alias, X.; Yang, J.S.; Ciampi, L.; Beltran-Sastre, V.; Torres-Méndez, A.; Irimia, M.; Schaefer, M.H.; Serrano, L. Silencing of SRRM4 Suppresses Microexon Inclusion and Promotes Tumor Growth across Cancers. PLoS Biol. 2021, 19, e3001138. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhou, J.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.S.; Li, L.; Zou, D.; Zhang, Y.; Jing, Z. Prosaposin Promotes the Proliferation and Tumorigenesis of Glioma through Toll-like Receptor 4 (TLR4)-Mediated NF-ΚB Signaling Pathway. EBioMedicine 2018, 37, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.W.; Didier, S.; Chan, A.; Paulino, V.; van Aelst, L.; Ruggieri, R.; Tran, N.L.; Byrne, A.T.; Symons, M. Guanine Nucleotide Exchange Factor Dock7 Mediates HGF-Induced Glioblastoma Cell Invasion via Rac Activation. Br. J. Cancer 2014, 110, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Labrecque, M.P.; Brown, L.G.; Coleman, I.M.; Lakely, B.; Brady, N.J.; Lee, J.K.; Nguyen, H.M.; Li, D.; Hanratty, B.; Haffner, M.C.; et al. RNA Splicing Factors SRRM3 and SRRM4 Distinguish Molecular Phenotypes of Castration-Resistant Neuroendocrine Prostate Cancer. Cancer Res. 2021, 81, 4736–4750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lucas, J.M.; Lam, H.M.; Dumpit, R.; Corey, E.; Chéry, L.; et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4698–4708. [Google Scholar] [CrossRef] [Green Version]
- Conn, V.M.; Gabryelska, M.; Marri, S.; Stringer, B.W.; Ormsby, R.J.; Penn, T.; Poonnoose, S.; Kichenadasse, G.; Conn, S.J. SRRM4 Expands the Repertoire of Circular RNAs by Regulating Microexon Inclusion. Cells 2020, 9, 2488. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Funayama, R.; Shirota, M.; Kikukawa, Y.; Ohira, M.; Karasawa, H.; Kobayashi, M.; Ohnuma, S.; Unno, M.; Nakayama, K. Alternative Microexon Splicing by RBFOX2 and PTBP1 Is Associated with Metastasis in Colorectal Cancer. Int. J. Cancer 2021, 149, 1787–1800. [Google Scholar] [CrossRef]
- Shi, X.; Singh, S.; Lin, E.; Li, H. Chimeric RNAs in Cancer. Adv. Clin. Chem. 2021, 100, 1–35. [Google Scholar]
- Shi, Y.; Yuan, J.; Rraklli, V.; Maxymovitz, E.; Cipullo, M.; Liu, M.; Li, S.; Westerlund, I.; Bedoya-Reina, O.C.; Bullova, P.; et al. Aberrant Splicing in Neuroblastoma Generates RNA-Fusion Transcripts and Provides Vulnerability to Spliceosome Inhibitors. Nucleic Acids Res. 2021, 49, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Paret, C.; Lehmann, N.; Bender, H.; Sprang, M.; Sommer, C.J.; Cana, D.; Seidmann, L.; Wingerter, A.; Neu, M.A.; el Malki, K.; et al. Identification of an Immunogenic Medulloblastoma-Specific Fusion Involving Epc2 and Gulp1. Cancers 2021, 13, 5838. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The Whole-Genome Landscape of Medulloblastoma Subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.W.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the Genomic Complexity Underlying Medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Sorana Morrissy, A.; Zichner, T.; Stútz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-Specific Structural Variation across 1000 Medulloblastoma Genomes. Nature 2012, 487, 49–56. [Google Scholar] [CrossRef]
- Luo, Z.; Dong, X.; Yu, J.; Xia, Y.; Berry, K.P.; Rao, R.; Xu, L.; Xue, P.; Chen, T.; Lin, Y.; et al. Genomic and Transcriptomic Analyses Reveals ZNF124 as a Critical Regulator in Highly Aggressive Medulloblastomas. Front. Cell Dev. Biol. 2021, 9, 634056. [Google Scholar] [CrossRef]
- Hong, E.M.; Ingemarsdotter, C.K.; Lever, A.M.L. Therapeutic Applications of Trans-Splicing. Br. Med. Bull. 2020, 136, 4–20. [Google Scholar] [CrossRef]
- Suñé-Pou, M.; Limeres, M.J.; Moreno-Castro, C.; Hernández-Munain, C.; Suñé-Negre, J.M.; Cuestas, M.L.; Suñé, C. Innovative Therapeutic and Delivery Approaches Using Nanotechnology to Correct Splicing Defects Underlying Disease. Front. Genet. 2020, 11, 731. [Google Scholar] [CrossRef]
- Mogilevsky, M.; Shimshon, O.; Kumar, S.; Mogilevsky, A.; Keshet, E.; Yavin, E.; Heyd, F.; Karni, R. Modulation of MKNK2 Alternative Splicing by Splice-Switching Oligonucleotides as a Novel Approach for Glioblastoma Treatment. Nucleic Acids Res. 2018, 46, 11396–11404. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Cheng, Y.; Zhang, C.; Chang, G.; Geng, X. A Novel Antisense Oligonucleotide Anchored on the Intronic Splicing Enhancer of HTERT Pre-MRNA Inhibits Telomerase Activity and Induces Apoptosis in Glioma Cells. J. Neuro-Oncol. 2019, 1, 57–68. [Google Scholar] [CrossRef]
- Braun, C.J.; Stanciu, M.; Boutz, P.L.; Patterson, J.C.; Calligaris, D.; Higuchi, F.; Neupane, R.; Fenoglio, S.; Cahill, D.P.; Wakimoto, H.; et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a Druggable Target for Glioblastoma Therapy. Neuro-Oncol. 2018, 20, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 Methylome Profiling Uncovers a Direct Link to Splicing Regulation in Acute Myeloid Leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Lin, H.; Luengo, J.I. Nucleoside Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. Bioorganic Med. Chem. Lett. 2019, 29, 1264–1269. [Google Scholar] [CrossRef]
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 Inhibition Disrupts Splicing and Stemness in Glioblastoma. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Visci, G.; Tolomeo, D.; Agostini, A.; Traversa, D.; Macchia, G.; Storlazzi, C.T. CircRNAs and Fusion-CircRNAs in Cancer: New Players in an Old Game. Cell. Signal. 2020, 75, 109747. [Google Scholar] [CrossRef] [PubMed]
- Rajappa, A.; Banerjee, S.; Sharma, V.; Khandelia, P. Circular RNAs: Emerging Role in Cancer Diagnostics and Therapeutics. Front. Mol. Biosci. 2020, 7, 256. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges MiR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity Through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef]
- Liu, X.; Abraham, J.M.; Cheng, Y.; Wang, Z.; Wang, Z.; Zhang, G.; Ashktorab, H.; Smoot, D.T.; Cole, R.N.; Boronina, T.N.; et al. Synthetic Circular RNA Functions as a MiR-21 Sponge to Suppress Gastric Carcinoma Cell Proliferation. Mol. Therapy. Nucleic Acids 2018, 13, 312–321. [Google Scholar] [CrossRef] [Green Version]
- Meganck, R.M.; Borchardt, E.K.; Castellanos Rivera, R.M.; Scalabrino, M.L.; Wilusz, J.E.; Marzluff, W.F.; Asokan, A. Tissue-Dependent Expression and Translation of Circular RNAs with Recombinant AAV Vectors In Vivo. Mol. Ther.-Nucleic Acids 2018, 13, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering Circular RNA for Potent and Stable Translation in Eukaryotic Cells. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.; Manley, J.; Lee, J.; Singh, S.R. The emerging roles of microRNAs in cancer metabolism. Cancer Lett. 2015, 356, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Woolf, E.C.; Syed, N.; Scheck, A.C. Tumor Metabolism, the Ketogenic Diet and β-Hydroxybutyrate: Novel Approaches to Adjuvant Brain Tumor Therapy. Front. Mol. Neurosci. 2016, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannataro, R.; Perri, R.; Gallelli, L.; Caroleo, M.C.; De Sarro, G.; Cione, E. Ketogenic Diet Acts on Body Remodeling and MicroRNAs Expression Profile. MicroRNA 2019, 8, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Han, S.R.; Lee, S.W. Therapeutic Applications of Group I Intron-Based Trans-Splicing Ribozymes. Wiley Interdiscip. Rev. RNA 2018, 9, e1466. [Google Scholar] [CrossRef] [PubMed]
- Puttaraju, M.; Jamison, S.F.; Mansfield, S.G.; Garcia-Blanco, M.A.; Mitchell, L.G. Spliceosome-Mediated RNA Trans-Splicing as a Tool for Gene Therapy. Nat. Biotechnol. 1999, 17, 246–252. [Google Scholar] [CrossRef]
- Mansfield, S.G.; Clark, R.H.; Puttaraju, M.; Kole, J.; Cohn, J.A.; Mitchell, L.G.; Garcia-Blanco, M.A. 5′ Exon Replacement and Repair by Spliceosome-Mediated RNA Trans-Splicing. RNA 2003, 9, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Sullenger, B.A. Induction of Wild-Type P53 Activity in Human Cancer Cells by Ribozymes That Repair Mutant P53 Transcripts. Proc. Natl. Acad. Sci. USA 2000, 97, 8490–8494. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.S.; Sullenger, B.A.; Lee, S.W. Ribozyme-Mediated Induction of Apoptosis in Human Cancer Cells by Targeted Repair of Mutant P53 RNA. Mol. Ther. 2004, 10, 365–372. [Google Scholar] [CrossRef]
- Kastanos, E.; Hjiantoniou, E.; Phylactou, L.A. Restoration of Protein Synthesis in Pancreatic Cancer Cells by Trans-Splicing Ribozymes. Biochem. Biophys. Res. Commun. 2004, 322, 930–934. [Google Scholar] [CrossRef]
- He, X.; Liu, F.; Yan, J.; Zhang, Y.; Yan, J.; Shang, H.; Dou, Q.; Zhao, Q.; Song, Y. Trans-Splicing Repair of Mutant P53 Suppresses the Growth of Hepatocellular Carcinoma Cells in Vitro and in Vivo. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, S.G.; Kole, J.; Puttaraju, M.; Yang, C.C.; Garcia-Blanco, M.A.; Cohn, J.A.; Mitchell, L.G. Repair of CFTR MRNA by Spliceosome-Mediated RNA Trans-Splicing. Gene Ther. 2000, 7, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, Q.; Mansfield, S.G.; Puttaraju, M.; Zhang, Y.; Zhou, W.; Cohn, J.A.; Garcia-Blanco, M.A.; Mitchell, L.G.; Engelhardt, J.F. Partial Correction of Endogenous ΔF508 CFTR in Human Cystic Fibrosis Airway Epithelia by Spliceosome-Mediated RNA Trans-Splicing. Nat. Biotechnol. 2002, 20, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, M.; Zhang, L.N.; Yan, Z.; Zak, R.; Ding, W.; Mansfield, S.G.; Mitchell, L.G.; Engelhardt, J.F. Spliceosome-Mediated RNA Trans-Splicing with Recombinant Adeno-Associated Virus Partially Restores Cystic Fibrosis Transmembrane Conductance Regulator Function to Polarized Human Cystic Fibrosis Airway Epithelial Cells. Hum. Gene Ther. 2005, 16, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Lou, H.H.; Boyer, J.L.; Limberis, M.P.; Vandenberghe, L.H.; Hackett, N.R.; Leopold, P.L.; Wilson, J.M.; Crystal, R.G. Functional Cystic Fibrosis Transmembrane Conductance Regulator Expression in Cystic Fibrosis Airway Epithelial Cells by AAV6.2-Mediated Segmental Trans-Splicing. Hum. Gene Ther. 2009, 20, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.; Mansfield, S.G.; Bartel, R.C.; Hiriyanna, S.; Mitchell, L.G.; Garcia-Blanco, M.A.; Walsh, C.E. Phenotype Correction of Hemophilia A Mice by Spliceosome-Mediated RNA Trans-Splicing. Nat. Med. 2003, 9, 1015–1019. [Google Scholar] [CrossRef]
- Kim, J.; Won, R.; Ban, G.; Ha Ju, M.; Sook Cho, K.; Young Han, S.; Jeong, J.S.; Lee, S.W. Targeted Regression of Hepatocellular Carcinoma by Cancer-Specific RNA Replacement through MicroRNA Regulation. Sci. Rep. 2015, 5, 12315. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Lee, S.W. Selective Expression of Transgene Using Hypoxia-Inducible Trans-Splicing Group I Intron Ribozyme. J. Biotechnol. 2014, 192, 22–27. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, K.T.; Lee, S.J.; Hong, S.H.; Moon, J.Y.; Yoon, E.K.; Kim, S.; Kim, E.O.; Kang, S.H.; Kim, S.K.; et al. Image-Aided Suicide Gene Therapy Utilizing Multifunctional HTERT-Targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016, 6, 357–368. [Google Scholar] [CrossRef]
- Song, M.S.; Jeong, J.S.; Ban, G.; Lee, J.H.; Won, Y.S.; Cho, K.S.; Kim, I.H.; Lee, S.W. Validation of Tissue-Specific Promoter-Driven Tumor-Targeting Trans-Splicing Ribozyme System as a Multifunctional Cancer Gene Therapy Device in Vivo. Cancer Gene Ther. 2009, 16, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; Jeong, J.S.; Lee, Y.J.; Jung, H.I.; Cho, K.S.; Kim, C.M.; Kwon, B.S.; Sullenger, B.A.; Lee, S.W.; Kim, I.H. In Vivo Reprogramming of HTERT by Trans-Splicing Ribozyme to Target Tumor Cells. Mol. Ther. 2008, 16, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Lee, S.W. Cancer-Selective Induction of Cytotoxicity by Tissue-Specific Expression of Targeted Trans-Splicing Ribozyme. FEBS Lett. 2006, 580, 5033–5043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, C.; Gratz, I.K.; Murauer, E.M.; Mayr, E.; Koller, U.; Bruckner-Tuderman, L.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Spliceosome-Mediated RNA Trans-Splicing Facilitates Targeted Delivery of Suicide Genes to Cancer Cells. Mol. Cancer Ther. 2011, 10, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero-Hinojosa, S.; Grant, M.; Panigrahi, A.; Zhang, H.; Caisova, V.; Bollard, C.M.; Rood, B.R. Proteogenomic Discovery of Neoantigens Facilitates Personalized Multi-Antigen Targeted T Cell Immunotherapy for Brain Tumors. Nat. Commun. 2021, 12, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alexandrov, P.N.; Jaber, V.; Lukiw, W.J. Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer’s Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7). Genes 2016, 7, 116. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| circRNAs | miRNAs | Dysregulation | Downstream Genes and Signaling Pathway Affected | Phenotype | Refs. |
|---|---|---|---|---|---|
| ciRS-7 | miR-7 | down | UBE2A | Neuropsychiatric-like phenotype | [53,54,235] |
| circNT5E | miR-422a | up | PI3K/AKT signaling | Proliferation, Invasion | [159] |
| circ0046701 | miR-142-3p | up | ITGB8 | Proliferation, Invasion | [160] |
| circHIPK3 | miR-654 | up | IGF2BP3 | Proliferation, Invasion | [55,161] |
| circ0014359 | miR-153 | up | PI3K/AKT signaling | Proliferation, migration, Invasion, apoptosis | [164] |
| circNFIX | miR-34a-5p | up | Notch signaling | Proliferation, migration, Invasion, apoptosis | [165] |
| circSHKBP1 | miR-544a miR-379 | up | FOXP1/FOXP2/AGG1 PI3K/AKT and ERK signaling | Proliferation, migration, angiogenesis | [166] |
| circ002136 | miR-138-5p | up | SOX13/SPON2 | Migration, invasion angiogenesis | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitolli, C.; Marini, A.; Sette, C.; Pagliarini, V. Non-Canonical Splicing and Its Implications in Brain Physiology and Cancer. Int. J. Mol. Sci. 2022, 23, 2811. https://doi.org/10.3390/ijms23052811
Pitolli C, Marini A, Sette C, Pagliarini V. Non-Canonical Splicing and Its Implications in Brain Physiology and Cancer. International Journal of Molecular Sciences. 2022; 23(5):2811. https://doi.org/10.3390/ijms23052811
Chicago/Turabian StylePitolli, Consuelo, Alberto Marini, Claudio Sette, and Vittoria Pagliarini. 2022. "Non-Canonical Splicing and Its Implications in Brain Physiology and Cancer" International Journal of Molecular Sciences 23, no. 5: 2811. https://doi.org/10.3390/ijms23052811
APA StylePitolli, C., Marini, A., Sette, C., & Pagliarini, V. (2022). Non-Canonical Splicing and Its Implications in Brain Physiology and Cancer. International Journal of Molecular Sciences, 23(5), 2811. https://doi.org/10.3390/ijms23052811