
A Possible Pathogenic PSEN2 Gly56Ser Mutation in a Korean Patient with Early-Onset Alzheimer’s Disease

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Mutational Analysis
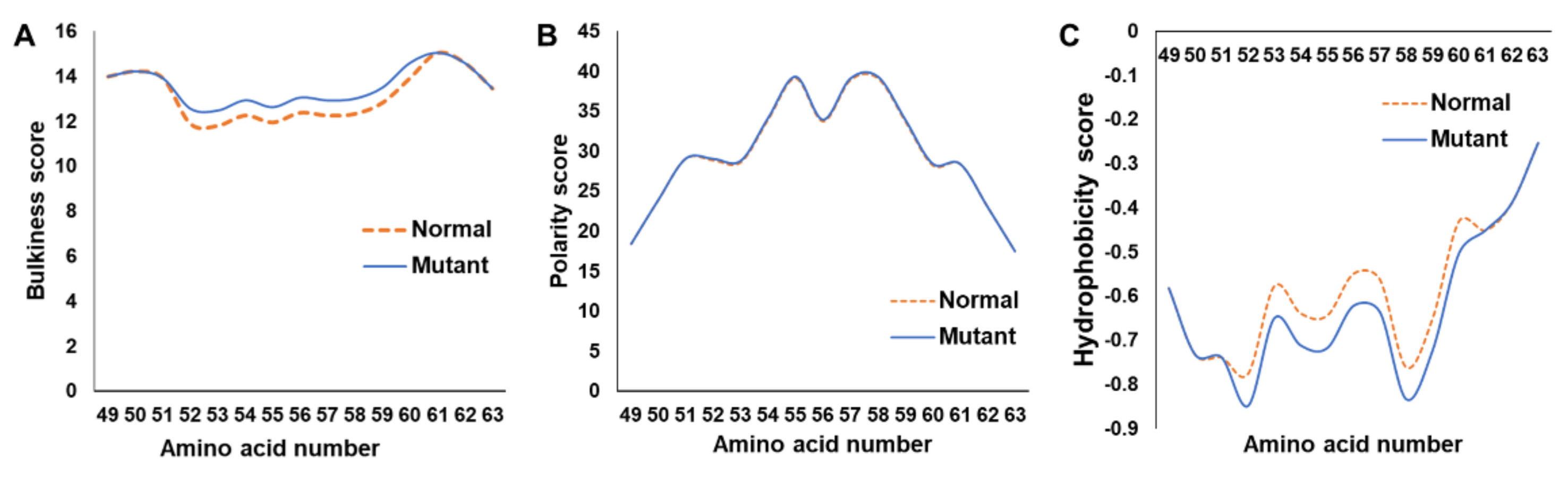
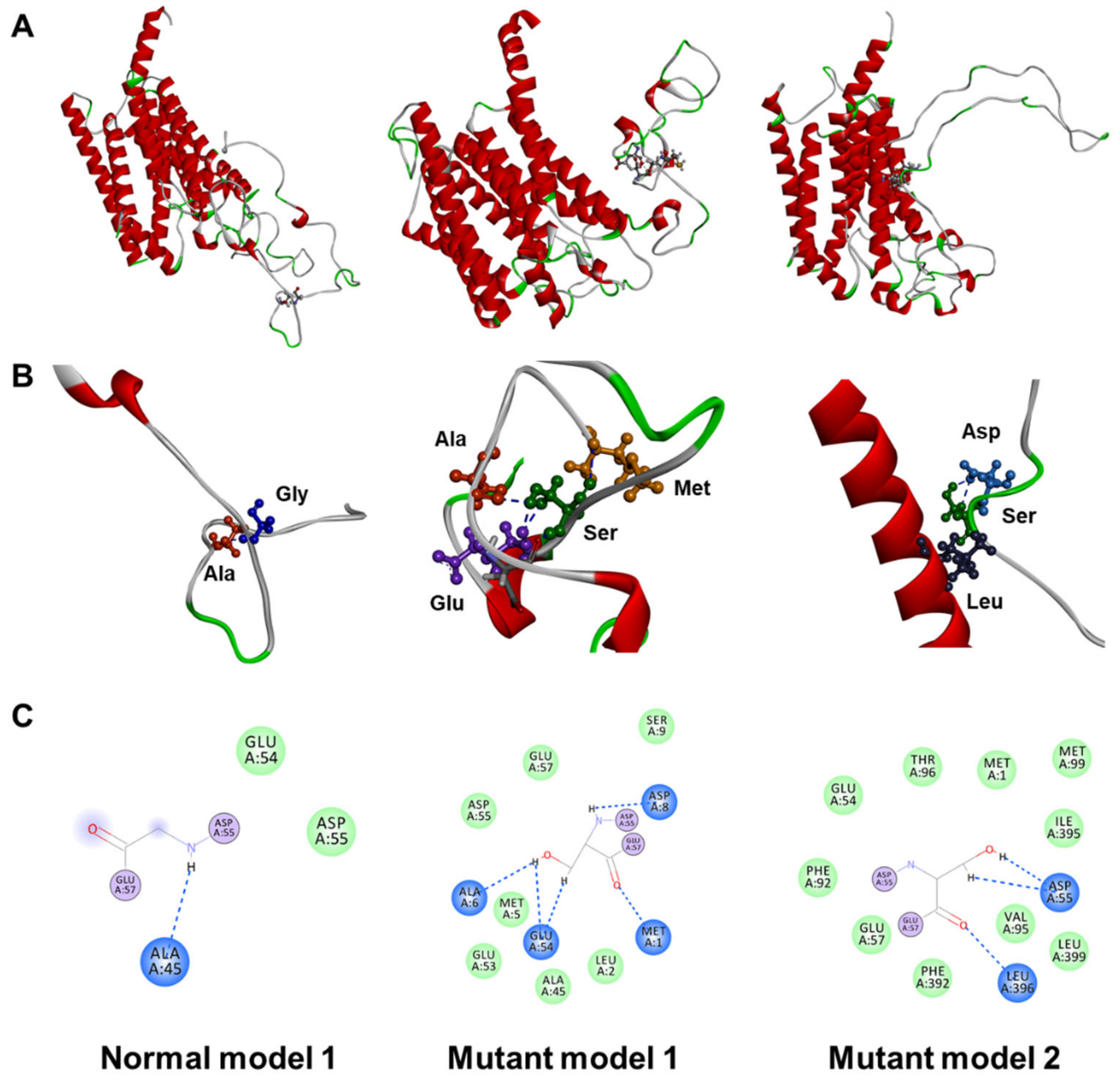
2.2. In Silico Assessment
3. Discussion
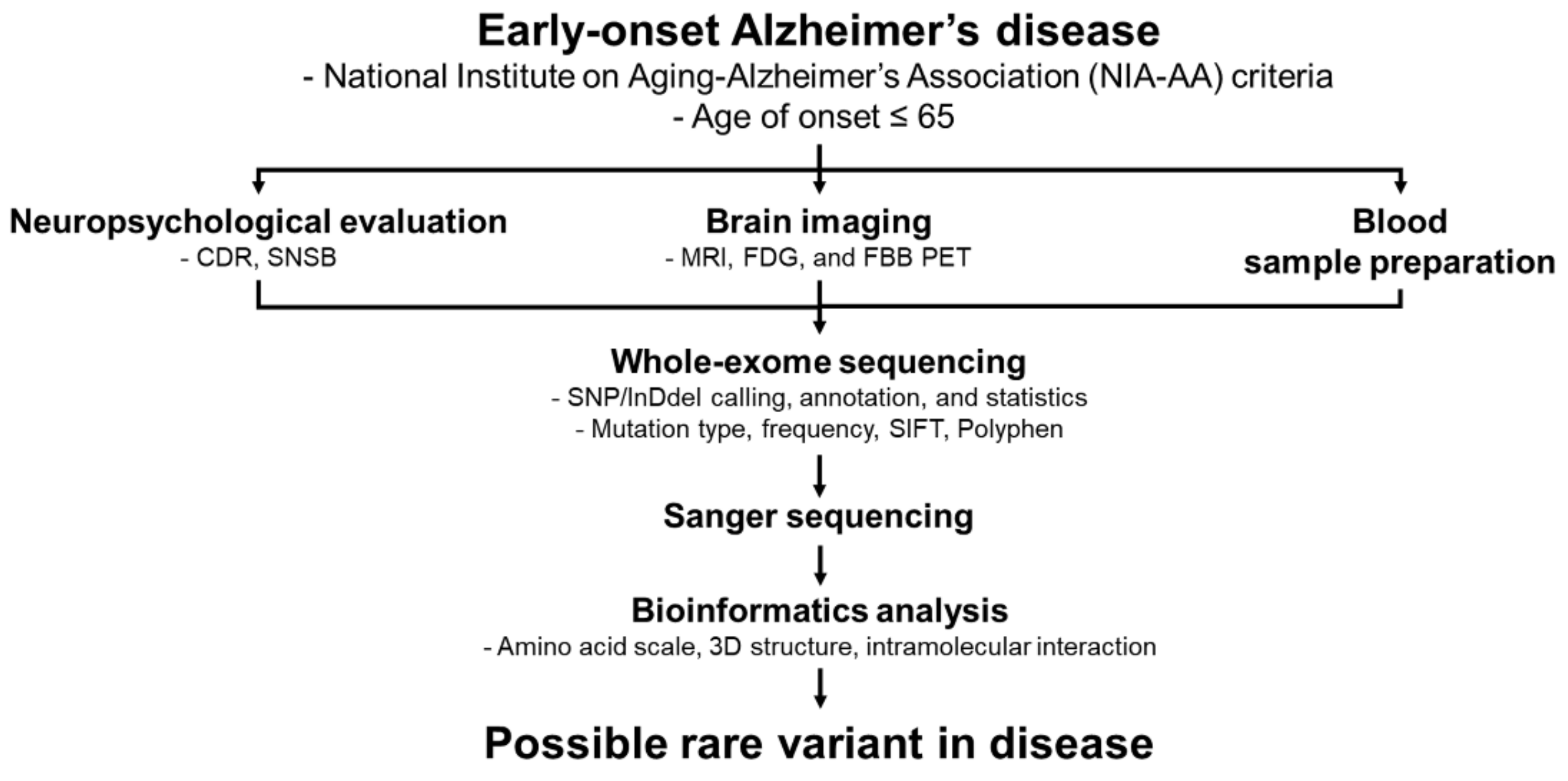
4. Materials and Methods
4.1. Patient Information
4.2. DNA Purification and Genetic Screening
4.3. In Silico Analyses
5. Conclusions
6. Limitations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate β-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front. Aging Neurosci. 2017, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci Rep. 2018, 8, 12592. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Lowe, V.J.; Senjem, M.L.; Weigand, S.D.; Kemp, B.J.; Shiung, M.M.; Knopman, D.S.; Boeve, B.F.; Klunk, W.E.; Mathis, C.A.; et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain 2008, 131, 665–680. [Google Scholar] [CrossRef] [Green Version]
- Falsetti, L.; Viticchi, G.; Zaccone, V.; Guerrieri, E.; Moroncini, G.; Luzzi, S.; Silvestrini, M. Shared Molecular Mechanisms among Alzheimer’s Disease, Neurovascular Unit Dysfunction and Vascular Risk Factors: A Narrative Review. Biomedicines 2022, 10, 439. [Google Scholar] [CrossRef]
- Mori, T.; Shimada, H.; Shinotoh, H.; Hirano, S.; Eguchi, Y.; Yamada, M.; Fukuhara, R.; Tanimukai, S.; Zhang, M.-R.; Kuwabara, S.; et al. Apathy correlates with prefrontal amyloid β deposition in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2014, 85, 449–455. [Google Scholar] [CrossRef]
- Garofalo, S.; Timmermann, C.; Battaglia, S.; Maier, M.E.; di Pellegrino, G. Mediofrontal Negativity Signals Unexpected Timing of Salient Outcomes. J. Cogn. Neurosci. 2017, 29, 718–727. [Google Scholar] [CrossRef]
- Cruciani, G.; Boccia, M.; Lingiardi, V.; Giovanardi, G.; Zingaretti, P.; Spitoni, G.F. An Exploratory Study on Resting-State Functional Connectivity in Individuals with Disorganized Attachment: Evidence for Key Regions in Amygdala and Hippocampus. Brain Sci. 2021, 11, 1539. [Google Scholar] [CrossRef]
- Khamassi, M.; Quilodran, R.; Enel, P.; Dominey, P.F.; Procyk, E. Behavioral Regulation and the Modulation of Information Coding in the Lateral Prefrontal and Cingulate Cortex. Cereb. Cortex 2014, 25, 3197–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzagalli, D.A.; Roberts, A.C. Prefrontal cortex and depression. Neuropsychopharmacology 2022, 47, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.H.; Brown, J.W. Hierarchical Error Representation: A Computational Model of Anterior Cingulate and Dorsolateral Prefrontal Cortex. Neural Comput. 2015, 27, 2354–2410. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G.; Starita, F. Revaluing the role of vmPFC in the acquisition of Pavlovian threat conditioning in humans. J. Neurosci. 2020, 40, 8491–8500. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The Genetics of Alzheimer Disease: Back to the Future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2021, 2021, 1–3. [Google Scholar] [CrossRef]
- Tashjian, S.M.; Zbozinek, T.D.; Mobbs, D. A Decision Architecture for Safety Computations. Trends Cogn. Sci. 2021, 25, 342–354. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Sciamanna, G.; Tortora, F.; Laricchiuta, D. Memories are not written in stone: Re-writing fear memories by means of non-invasive brain stimulation and optogenetic manipulations. Neurosci. Biobehav. Rev. 2021, 127, 334–352. [Google Scholar] [CrossRef]
- Battaglia, S. Neurobiological advances of learned fear in humans. Adv. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horré, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456.e414. [Google Scholar] [CrossRef] [Green Version]
- Giau, V.V.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. APP, PSEN1, and PSEN2 Mutations in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 2019, 20, 4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, N.S.; Rossor, M.N. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark. Med. 2010, 4, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilotto, A.; Padovani, A.; Borroni, B. Clinical, Biological, and Imaging Features of Monogenic Alzheimer’s Disease. BioMed Res. Int. 2013, 2013, 689591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, N.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of Alzheimer’s disease: An update. Ann. Med. 2008, 40, 562–583. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; Lau, P.; Troakes, C.; Deramecourt, V.; Gele, P.; Van Loo, P.; Voet, T.; De Strooper, B. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimers Dement. 2015, 11, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Perrone, F.; Bjerke, M.; Hens, E.; Sieben, A.; Timmers, M.; De Roeck, A.; Vandenberghe, R.; Sleegers, K.; Martin, J.J.; De Deyn, P.P.; et al. Amyloid-β(1-43) cerebrospinal fluid levels and the interpretation of APP, PSEN1 and PSEN2 mutations. Alzheimers Res. Ther. 2020, 12, 108. [Google Scholar] [CrossRef]
- Sleegers, K.; Roks, G.; Theuns, J.; Aulchenko, Y.S.; Rademakers, R.; Cruts, M.; van Gool, W.A.; Van Broeckhoven, C.; Heutink, P.; Oostra, B.A.; et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain 2004, 127, 1641–1649. [Google Scholar] [CrossRef]
- Hsu, S.; Pimenova, A.A.; Hayes, K.; Villa, J.A.; Rosene, M.J.; Jere, M.; Goate, A.M.; Karch, C.M. Systematic validation of variants of unknown significance in APP, PSEN1 and PSEN2. Neurobiol. Dis. 2020, 139, 104817. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Baquero, M.; Blesa, R.; Boada, M.; Brás, J.M.; Bullido, M.J.; Calado, A.; Crook, R.; Ferreira, C.; Frank, A.; et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging 2010, 31, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Koriath, C.; Kenny, J.; Adamson, G.; Druyeh, R.; Taylor, W.; Beck, J.; Quinn, L.; Mok, T.H.; Dimitriadis, A.; Norsworthy, P.; et al. Predictors for a dementia gene mutation based on gene-panel next-generation sequencing of a large dementia referral series. Mol. Psychiatry 2020, 25, 3399–3412. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatto, E.M.; Rojas, G.J.; Nemirovsky, S.I.; Da Prat, G.; Persi, G.; Cesarini, M.; Etcheverry, J.L.; Rojas, N.G.; Parisi, V.; Cordoba, M.; et al. A novel mutation in PSEN1 (p.Arg41Ser) in an Argentinian woman with early onset Parkinsonism. Parkinsonism Relat. Disord. 2020, 77, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Tampi, R.R.; Forester, B.P.; Agronin, M. Aducanumab: Evidence from clinical trial data and controversies. Drugs Context 2021, 10, 2021-7-3. [Google Scholar] [CrossRef]
- Buss, S.S.; Fried, P.J.; Pascual-Leone, A. Therapeutic noninvasive brain stimulation in Alzheimer’s disease and related dementias. Curr. Opin. Neurol. 2019, 32, 292–304. [Google Scholar] [CrossRef]
- Patrick, R.; Lê Cao, K.-A.; Kobe, B.; Bodén, M. PhosphoPICK: Modelling cellular context to map kinase-substrate phosphorylation events. Bioinformatics 2014, 31, 382–389. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation | Age of Onset (y) | Family History | Functional Data | Clinical Phenotype |
|---|---|---|---|---|---|
| PSEN1 | Asn24Ser | 65.8 | Familial (five carriers with AD from China) | Damaging effect predicted by at least two algorithms | AD |
| Asn39Tyr | Not available | Not available | Conflicting results from in silico predictive analyses | EOAD | |
| Arg41Ser | 35 | No family history | Cerebrospinal fluid levels of Aβ and tau were normal; phospho-tau levels were elevated | EOPD | |
| PSEN2 | Gly34Ser | 61.6 (China) | Familial (three families and ten carriers with AD or MCI from China | Unchanged Aβ42/Aβ40 ratio (The Netherlands) | LOAD (The Netherlands) EOAD and LOAD (China) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, K.-H.; Kang, M.-J.; Bae, H.; Kim, D.; Park, J.; An, S.-S.A.; Jeong, D.-E. A Possible Pathogenic PSEN2 Gly56Ser Mutation in a Korean Patient with Early-Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2967. https://doi.org/10.3390/ijms23062967
Shim K-H, Kang M-J, Bae H, Kim D, Park J, An S-SA, Jeong D-E. A Possible Pathogenic PSEN2 Gly56Ser Mutation in a Korean Patient with Early-Onset Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(6):2967. https://doi.org/10.3390/ijms23062967
Chicago/Turabian StyleShim, Kyu-Hwan, Min-Ju Kang, Heewon Bae, Danyeong Kim, Jiwon Park, Seong-Soo A. An, and Da-Eun Jeong. 2022. "A Possible Pathogenic PSEN2 Gly56Ser Mutation in a Korean Patient with Early-Onset Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 6: 2967. https://doi.org/10.3390/ijms23062967
APA StyleShim, K.-H., Kang, M.-J., Bae, H., Kim, D., Park, J., An, S.-S. A., & Jeong, D.-E. (2022). A Possible Pathogenic PSEN2 Gly56Ser Mutation in a Korean Patient with Early-Onset Alzheimer’s Disease. International Journal of Molecular Sciences, 23(6), 2967. https://doi.org/10.3390/ijms23062967