
Optimized Protocol for Proportionate CNS Cell Retrieval as a Versatile Platform for Cellular and Molecular Phenomapping in Aging and Neurodegeneration

 , and
, and
Abstract
:
1. Introduction
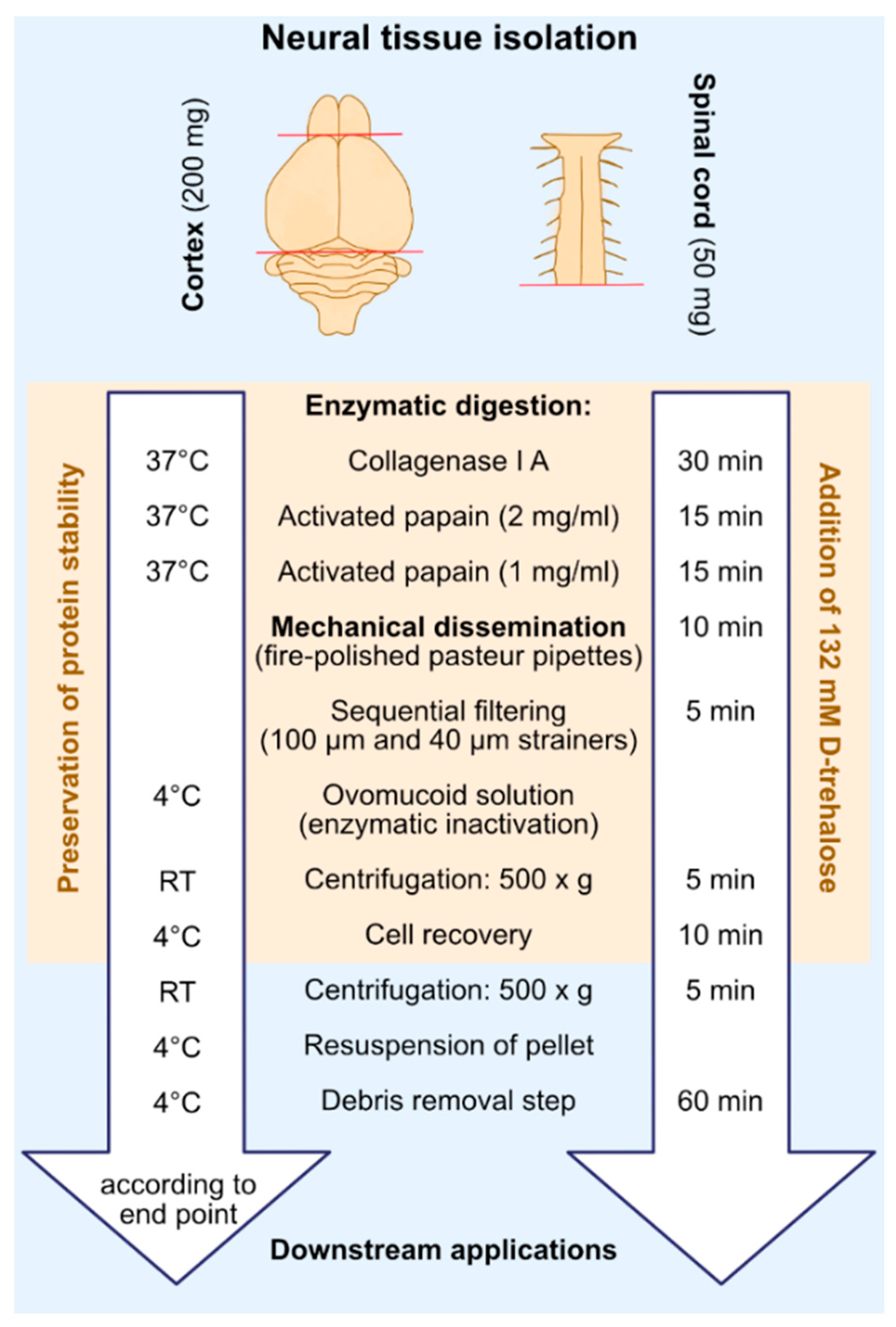
2. Results
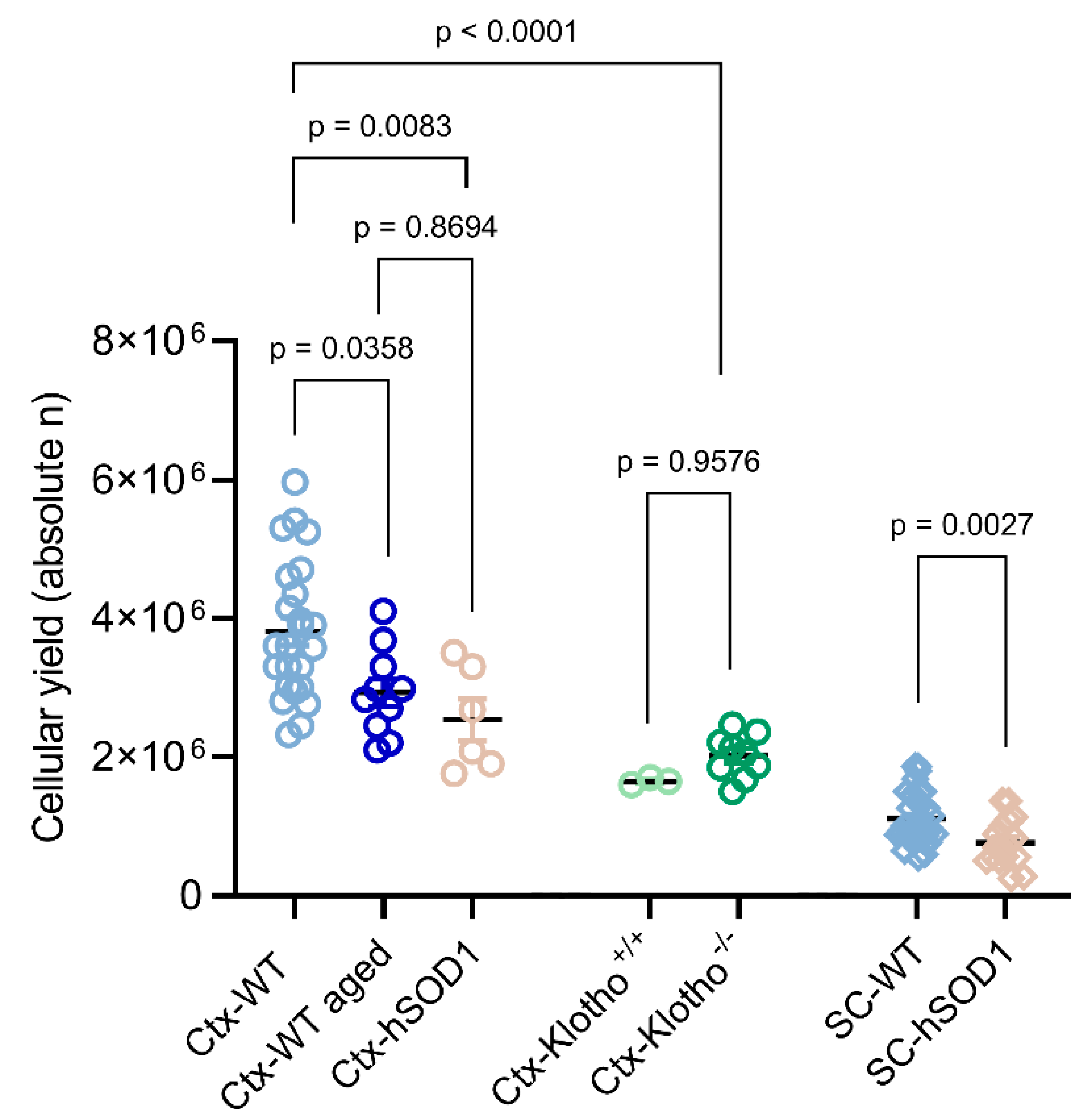
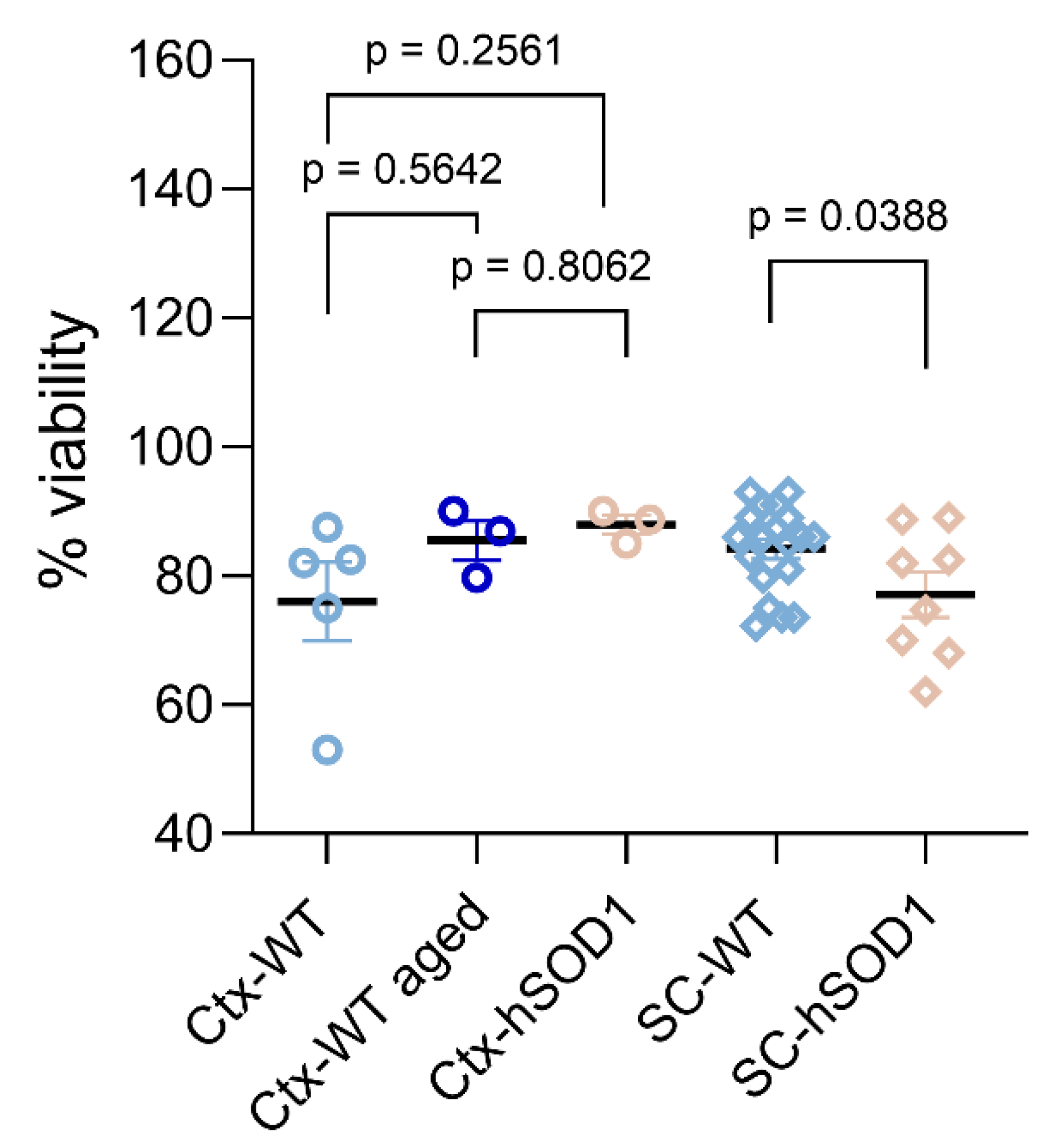
2.1. Quality Assessment of Cell Isolates
2.1.1. Total Cellular Yield
2.1.2. Viability of Isolated Cells
2.1.3. Preservation of Physiological Proportions of Neural Cell Populations
Cell Proportions for Spinal Cord Isolates
Cell Proportions for Cortical Isolates
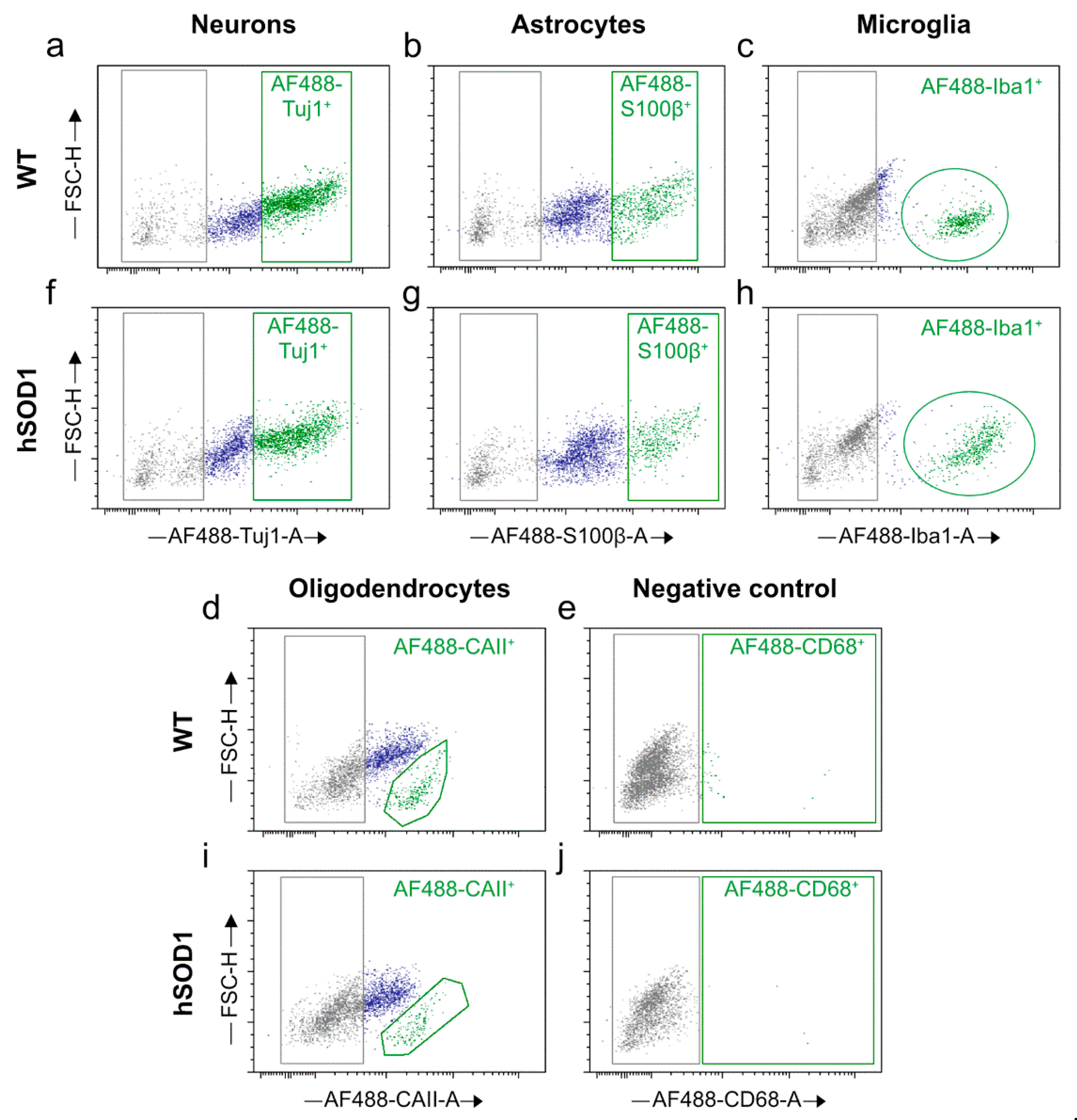

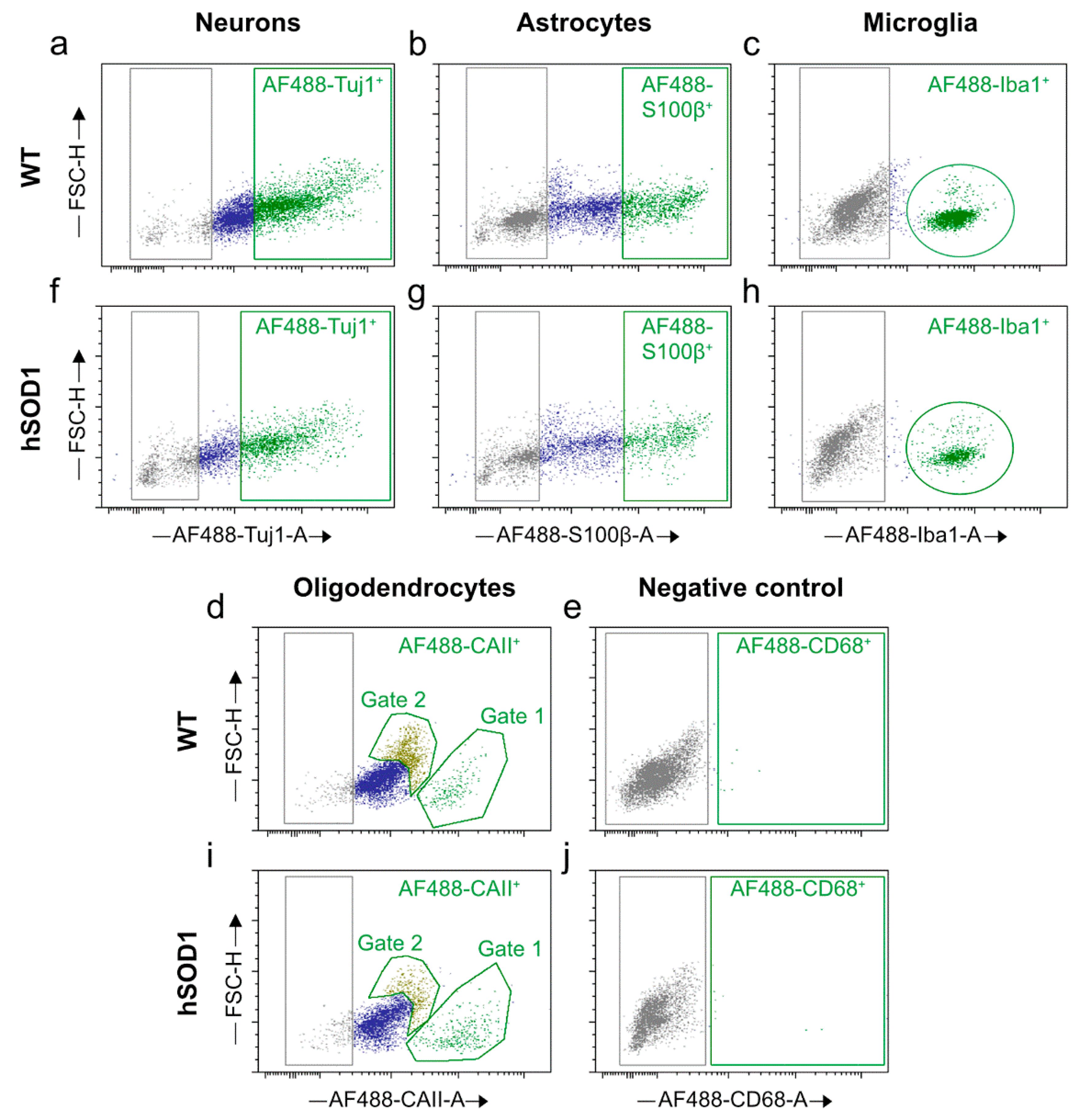
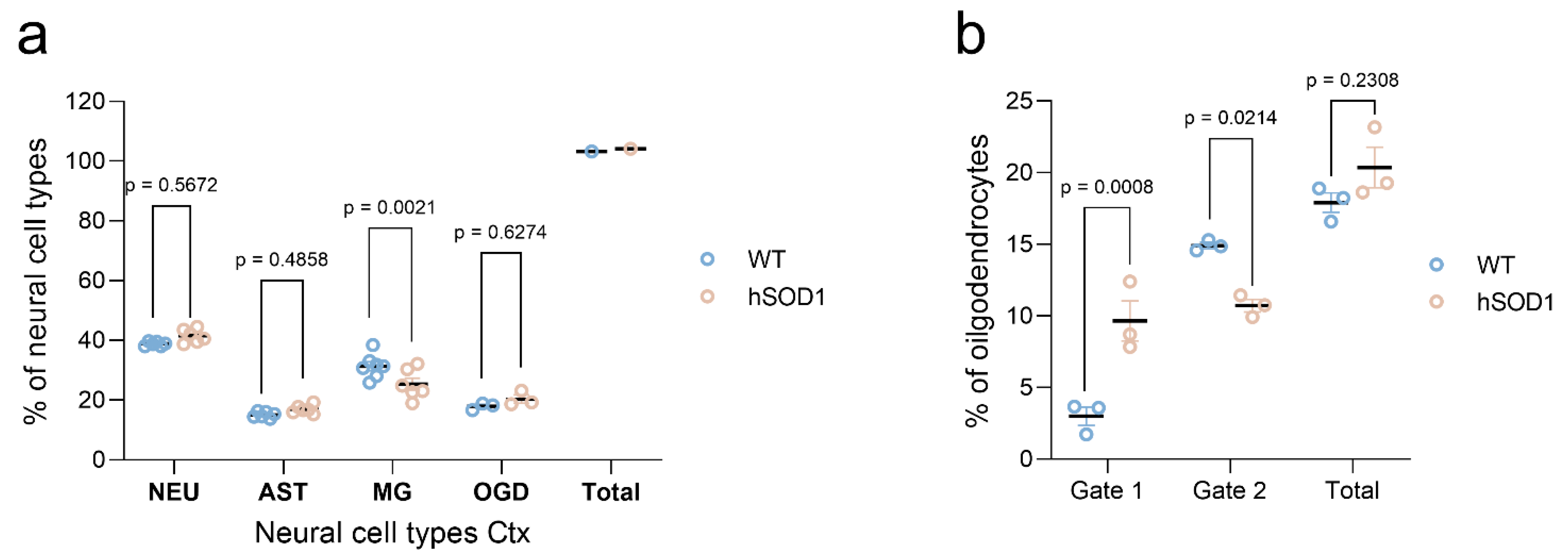
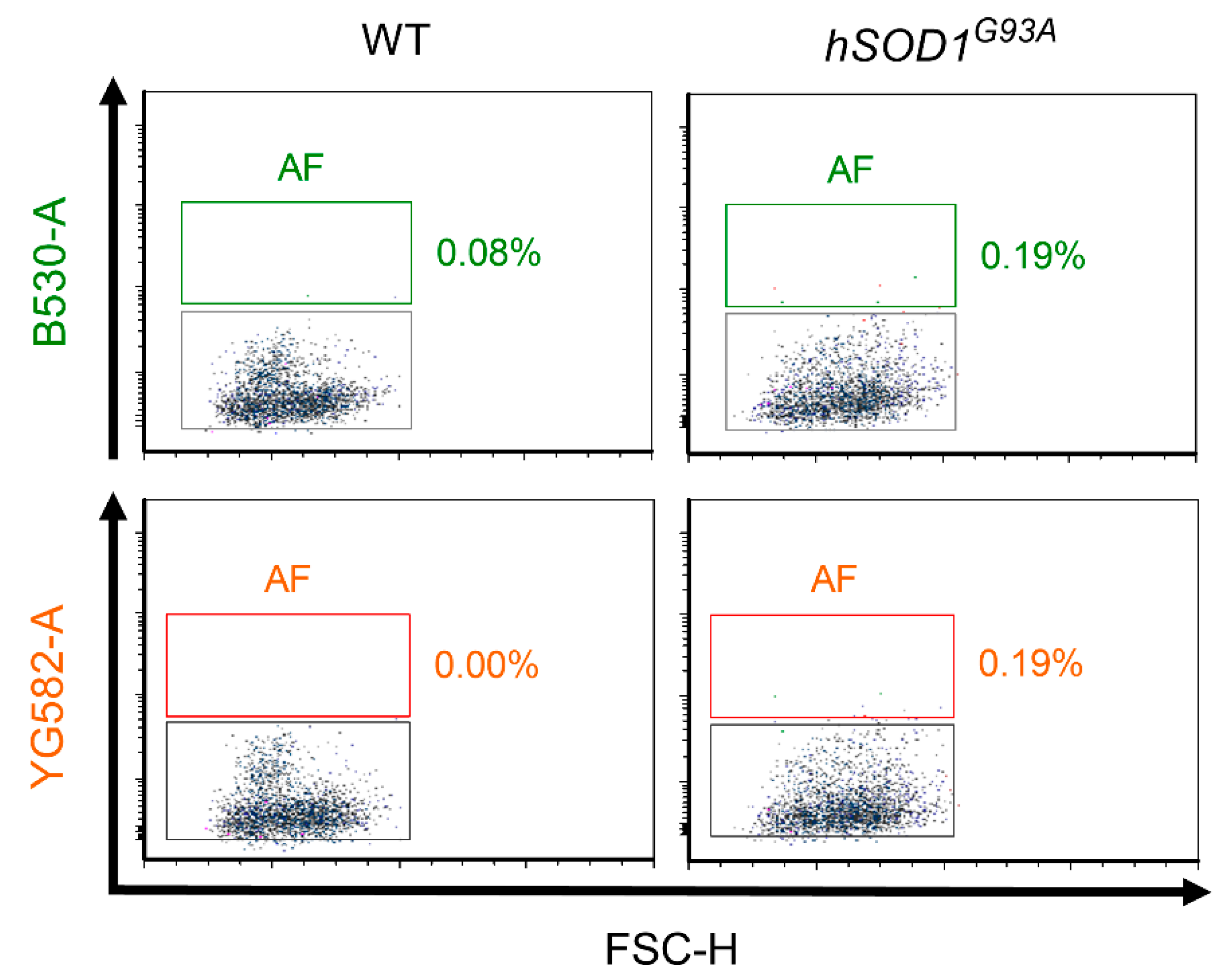
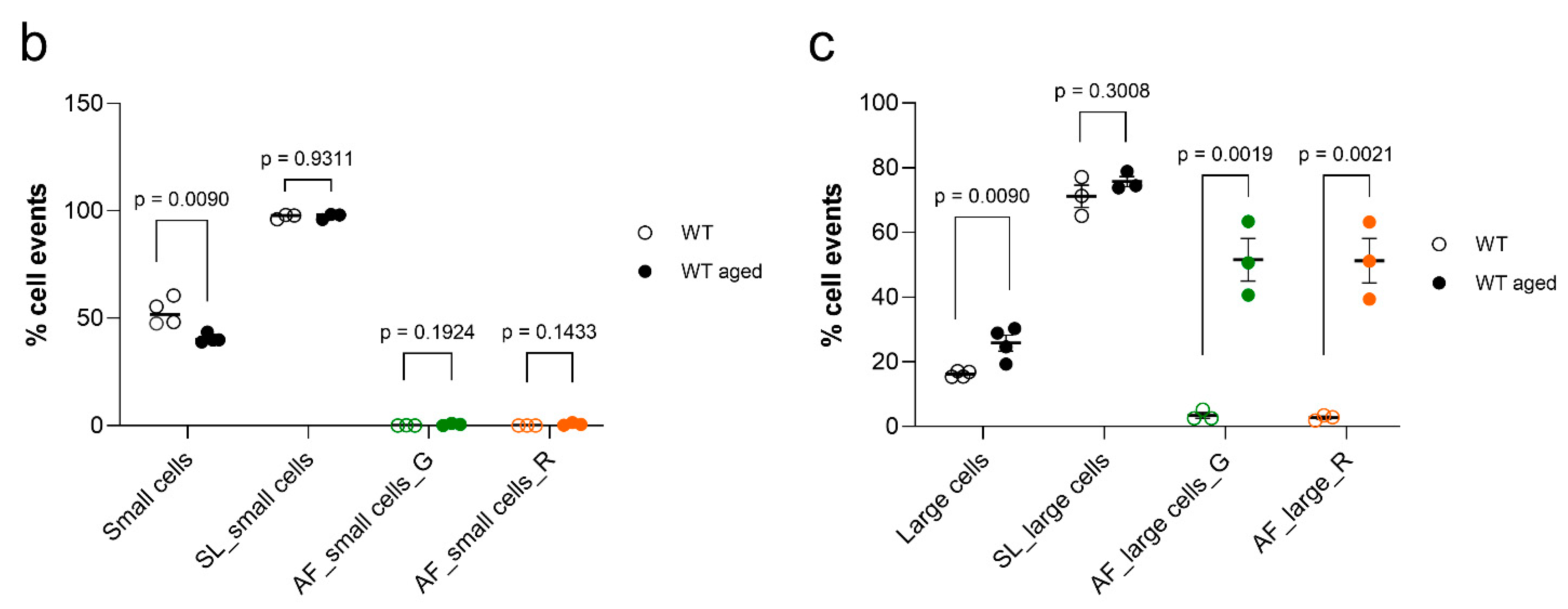
2.2. Autofluorescence Pattern in Young, Aged, Progeroid, and Neurodegenerative Specimens
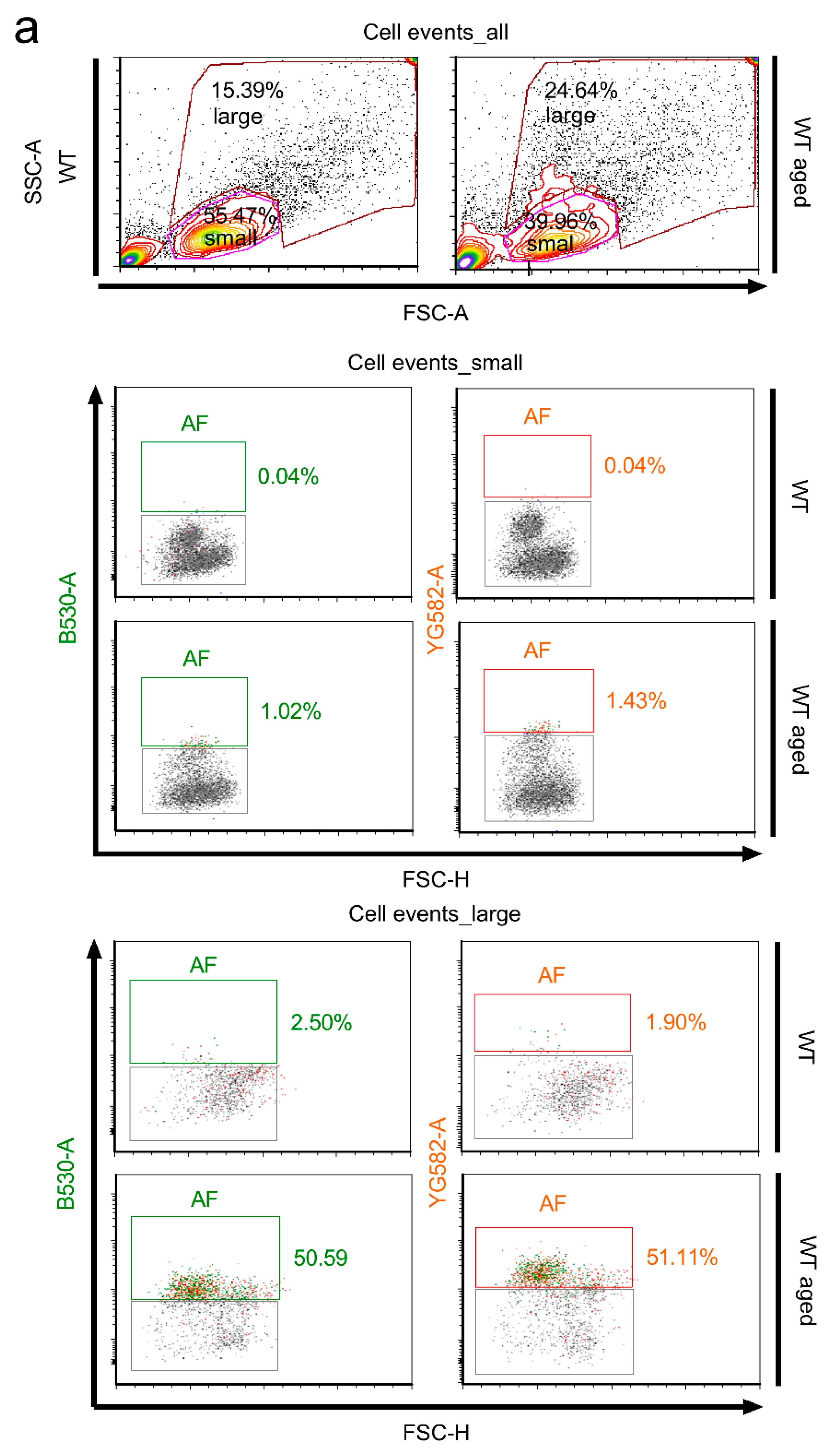
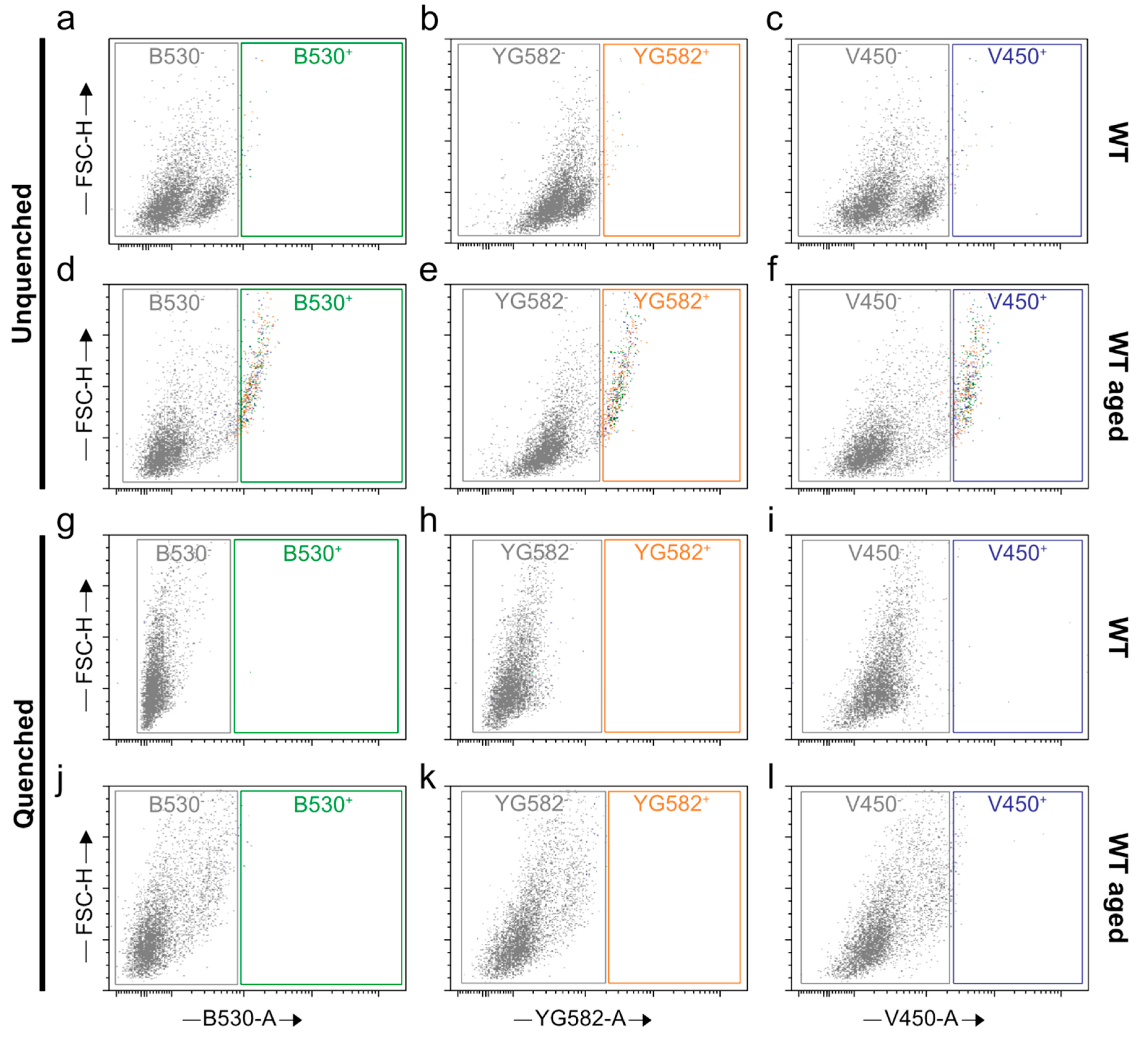
2.3. Stratification of Cells Responsible for Enhanced Autofluorescence in Highly Aged Specimens
2.4. Quenching of Autofluorescence
3. Discussion
4. Materials and Methods
4.1. Acquisition of Biological Material
4.2. Animal Anesthesia and Perfusion
4.3. Extraction of Cortex and Spinal Cord Tissue
4.4. Digestion of Connective Tissue
4.5. Neural Cell Isolation
4.6. Debris Removal Step
4.7. Fixation of the Isolated Neural Cells
4.8. Quenching of Unspecific Fluorescence Signals
4.9. Immunostaining for Flow Cytometry-Based Separation of Neural Cell Entities
4.10. Flow Cytometry-Based Assessment of Different Neural Cell Proportions

4.11. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brewer, G.J.; Torricelli, J.R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2007, 2, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, J.A.; Soriano, E. Regenerating cortical connections in a dish: The entorhino-hippocampal organotypic slice co-culture as tool for pharmacological screening of molecules promoting axon regeneration. Nat. Protoc. 2010, 5, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giandomenico, S.L.; Sutcliffe, M.; Lancaster, M.A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 2021, 16, 579–602. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.; Marticke, J.K.; Happold, C.J.; Schmeer, C.; Isenmann, S. A primary culture technique of adult retina for regeneration studies on adult CNS neurons. Nat. Protoc. 2007, 2, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Matsuki, N.; Ikegaya, Y. Targeted axon-attached recording with fluorescent patch-clamp pipettes in brain slices. Nat. Protoc. 2012, 7, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Magrane, J. Isolation and culture of neurons and astrocytes from the mouse brain cortex. Methods Mol. Biol. 2011, 793, 63–75. [Google Scholar] [PubMed]
- Sciarretta, C.; Minichiello, L. The preparation of primary cortical neuron cultures and a practical application using immunofluorescent cytochemistry. Methods Mol. Biol. 2010, 633, 221–231. [Google Scholar] [PubMed]
- Wiese, S.; Herrmann, T.; Drepper, C.; Jablonka, S.; Funk, N.; Klausmeyer, A.; Rogers, M.L.; Rush, R.; Sendtner, M. Isolation and enrichment of embryonic mouse motoneurons from the lumbar spinal cord of individual mouse embryos. Nat. Protoc. 2010, 5, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, G.M., 3rd; Lee, S.H.; Singh, D.; Yuan, Y.; Ng, Y.G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.K.; Karsten, S.L.; Gray, M.; Geschwind, D.H.; Yang, X.W. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat. Neurosci. 2006, 9, 443–452. [Google Scholar] [CrossRef]
- Schroeter, C.B.; Herrmann, A.M.; Bock, S.; Vogelsang, A.; Eichler, S.; Albrecht, P.; Meuth, S.G.; Ruck, T. One Brain-All Cells: A Comprehensive Protocol to Isolate All Principal CNS-Resident Cell Types from Brain and Spinal Cord of Adult Healthy and EAE Mice. Cells 2021, 10, 651. [Google Scholar] [CrossRef] [PubMed]
- Foo, L.C.; Allen, N.J.; Bushong, E.A.; Ventura, P.B.; Chung, W.S.; Zhou, L.; Cahoy, J.D.; Daneman, R.; Zong, H.; Ellisman, M.H.; et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 2011, 71, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Beaudet, M.J.; Yang, Q.; Cadau, S.; Blais, M.; Bellenfant, S.; Gros-Louis, F.; Berthod, F. High yield extraction of pure spinal motor neurons, astrocytes and microglia from single embryo and adult mouse spinal cord. Sci. Rep. 2015, 5, 16763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.Y.; Li, X.T.; Peng, S.W.; Xiao, J.F.; Liu, C.; Fang, G.; Chen, K.C.; Chen, G.Q. The behaviour of neural stem cells on polyhydroxyalkanoate nanofiber scaffolds. Biomaterials 2010, 31, 3967–3975. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.P.; Dougherty, J.D.; Heiman, M.; Schmidt, E.F.; Stevens, T.R.; Ma, G.; Bupp, S.; Shrestha, P.; Shah, R.D.; Doughty, M.L.; et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 2008, 135, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiman, M.; Schaefer, A.; Gong, S.; Peterson, J.D.; Day, M.; Ramsey, K.E.; Suarez-Farinas, M.; Schwarz, C.; Stephan, D.A.; Surmeier, D.J.; et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell 2008, 135, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallos, J.; Jeng, S.; McWeeney, S.; Martin, I. Dopamine neuron-specific LRRK2 G2019S effects on gene expression revealed by translatome profiling. Neurobiol. Dis. 2021, 155, 105390. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Imeri, M.; Schwarz, P.; Aguzzi, A. Ribosomal profiling during prion disease uncovers progressive translational derangement in glia but not in neurons. eLife 2020, 9, e62911. [Google Scholar] [CrossRef] [PubMed]
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillip, J.M.; Wu, P.H.; Gilkes, D.M.; Williams, W.; McGovern, S.; Daya, J.; Chen, J.; Aifuwa, I.; Lee, J.S.H.; Fan, R.; et al. Biophysical and biomolecular determination of cellular age in humans. Nat. Biomed. Eng. 2017, 1, 93. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Herrera, F.; Taoro-Gonzalez, L.; Valdes-Baizabal, C.; Diaz, M.; Marin, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Jin, H.K.; Bae, J.S. Potential therapeutic target for aging and age-related neurodegenerative diseases: The role of acid sphingomyelinase. Exp. Mol. Med. 2020, 52, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M. The genetics and neuropathology of neurodegenerative disorders: Perspectives and implications for research and clinical practice. Acta Neuropathol. 2012, 124, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Burtner, C.R.; Kennedy, B.K. Progeria syndromes and ageing: What is the connection? Nat. Rev. Mol. Cell Biol. 2010, 11, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Carrero, D.; Soria-Valles, C.; Lopez-Otin, C. Hallmarks of progeroid syndromes: Lessons from mice and reprogrammed cells. Dis. Model. Mech. 2016, 9, 719–735. [Google Scholar] [CrossRef] [Green Version]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Wan, T.; Miwa, S. Senescence in Post-Mitotic Cells: A Driver of Aging? Antioxid. Redox Signal. 2021, 34, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kumar, D.; Jha, N.K.; Jha, S.K.; Ambasta, R.K.; Kumar, P. Re-expression of cell cycle markers in aged neurons and muscles: Whether cells should divide or die? Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Wengerodt, D.; Schmeer, C.; Witte, O.W.; Kretz, A. Amitosenescence and Pseudomitosenescence: Putative New Players in the Aging Process. Cells 2019, 8, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, J.; Wang, B.; Brandenburg, S.M.; Basisty, N.; Evangelou, K.; Varela-Eirin, M.; Campisi, J.; Schilling, B.; Gorgoulis, V.; Demaria, M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 2021, 16, 2471–2498. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S.; Watson, C.; Paxinos, G. Distribution of neurons in functional areas of the mouse cerebral cortex reveals quantitatively different cortical zones. Front. Neuroanat. 2013, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Yoon, G.; Kang, H.T. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 2009, 66, 2503–2524. [Google Scholar] [CrossRef]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brustle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef]
- Guez-Barber, D.; Fanous, S.; Harvey, B.K.; Zhang, Y.; Lehrmann, E.; Becker, K.G.; Picciotto, M.R.; Hope, B.T. FACS purification of immunolabeled cell types from adult rat brain. J. Neurosci. Methods 2012, 203, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Wagatsuma, A.; Noro, Y.; Kuji, T.; Asaka-Oba, A.; Watahiki, A.; Gurnot, C.; Fagiolini, M.; Hensch, T.K.; Carninci, P. Trehalose-enhanced isolation of neuronal sub-types from adult mouse brain. Biotechniques 2012, 52, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Sloane, J.A.; Li, H.; Aytan, N.; Giannaris, E.L.; Zeldich, E.; Hinman, J.D.; Dedeoglu, A.; Rosene, D.L.; Bansal, R.; et al. The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J. Neurosci. 2013, 33, 1927–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eide, L.; McMurray, C.T. Culture of adult mouse neurons. Biotechniques 2005, 38, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ain, Q.; Schmeer, C.; Penndorf, D.; Fischer, M.; Bondeva, T.; Forster, M.; Haenold, R.; Witte, O.W.; Kretz, A. Cell cycle-dependent and -independent telomere shortening accompanies murine brain aging. Aging 2018, 10, 3397–3420. [Google Scholar] [CrossRef] [PubMed]
- Penndorf, D.; Tadic, V.; Witte, O.W.; Grosskreutz, J.; Kretz, A. DNA strand breaks and TDP-43 mislocation are absent in the murine hSOD1G93A model of amyotrophic lateral sclerosis in vivo and in vitro. PLoS ONE 2017, 12, e0183684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floriddia, E.M.; Lourenco, T.; Zhang, S.; van Bruggen, D.; Hilscher, M.M.; Kukanja, P.; Goncalves Dos Santos, J.P.; Altinkok, M.; Yokota, C.; Llorens-Bobadilla, E.; et al. Distinct oligodendrocyte populations have spatial preference and different responses to spinal cord injury. Nat. Commun. 2020, 11, 5860. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.B.; Jackiewicz, M.; McKenzie, I.A.; Kougioumtzidou, E.; Grist, M.; Richardson, W.D. Remarkable Stability of Myelinating Oligodendrocytes in Mice. Cell Rep. 2017, 21, 316–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nott, A.; Schlachetzki, J.C.M.; Fixsen, B.R.; Glass, C.K. Nuclei isolation of multiple brain cell types for omics interrogation. Nat. Protoc. 2021, 16, 1629–1646. [Google Scholar] [CrossRef]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Bertolo, A.; Baur, M.; Guerrero, J.; Potzel, T.; Stoyanov, J. Autofluorescence is a Reliable in vitro Marker of Cellular Senescence in Human Mesenchymal Stromal Cells. Sci. Rep. 2019, 9, 2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; Heiland, D.H.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, A.; Russo, M.J.; Bieganski, R.; Fowler, A.; Cheley, S.; Bayley, H.; Toner, M. Intracellular trehalose improves the survival of cryopreserved mammalian cells. Nat. Biotechnol. 2000, 18, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, A.; Bailey, S.E.; Toner, M.; Toth, T.L. Successful cryopreservation of mouse oocytes by using low concentrations of trehalose and dimethylsulfoxide. Biol. Reprod. 2009, 80, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.Y.; Tan, P.C.; Xie, Y.; Zhang, X.J.; Zhang, P.Q.; Gao, Y.M.; Zhou, S.B.; Li, Q.F. The combination of trehalose and glycerol: An effective and non-toxic recipe for cryopreservation of human adipose-derived stem cells. Stem Cell Res. Ther. 2020, 11, 460. [Google Scholar] [CrossRef]
- Jain, N.K.; Roy, I. Effect of trehalose on protein structure. Protein Sci. 2009, 18, 24–36. [Google Scholar] [CrossRef]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Zhang, M.; Oldenhof, H.; Sydykov, B.; Bigalk, J.; Sieme, H.; Wolkers, W.F. Freeze-drying of mammalian cells using trehalose: Preservation of DNA integrity. Sci. Rep. 2017, 7, 6198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinaga, K.; Yoshioka, H.; Kurosaki, H.; Hirasawa, M.; Uritani, M.; Hasegawa, K. Protection by trehalose of DNA from radiation damage. Biosci. Biotechnol. Biochem. 1997, 61, 160–161. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Fielder, E.; von Zglinicki, T.; Jurk, D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J. Alzheimers Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Salmonowicz, H.; Gladyshev, V.N. Integrating cellular senescence with the concept of damage accumulation in aging: Relevance for clearance of senescent cells. Aging Cell 2019, 18, e12841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Husseman, J.W.; Nochlin, D.; Vincent, I. Mitotic activation: A convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol. Aging 2000, 21, 815–828. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef] [Green Version]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S.; et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology 2014, 15, 47–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chio, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Dewan, R.; Chia, R.; Ding, J.; Hickman, R.A.; Stein, T.D.; Abramzon, Y.; Ahmed, S.; Sabir, M.S.; Portley, M.K.; Tucci, A.; et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron 2021, 109, 448–460.e4. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojiljkovic, M.R.; Ain, Q.; Bondeva, T.; Heller, R.; Schmeer, C.; Witte, O.W. Phenotypic and functional differences between senescent and aged murine microglia. Neurobiol. Aging 2019, 74, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Shiozaki, M.; Yoshimura, K.; Shibata, M.; Koike, M.; Matsuura, N.; Uchiyama, Y.; Gotow, T. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience 2008, 152, 924–941. [Google Scholar] [CrossRef]
- Duce, J.A.; Podvin, S.; Hollander, W.; Kipling, D.; Rosene, D.L.; Abraham, C.R. Gene profile analysis implicates Klotho as an important contributor to aging changes in brain white matter of the rhesus monkey. Glia 2008, 56, 106–117. [Google Scholar] [CrossRef]
- Leibrock, C.B.; Voelkl, J.; Kuro, O.M.; Lang, F.; Lang, U.E. 1,25(OH)2D3 dependent overt hyperactivity phenotype in klotho-hypomorphic mice. Sci. Rep. 2016, 6, 24879. [Google Scholar] [CrossRef] [Green Version]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Fujimori, T.; Nabeshima, Y. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology 2002, 143, 683–689. [Google Scholar] [CrossRef] [PubMed]
- De Novo FCS Express; Version 5 Plus; De Novo Software: Pasadena, CA, USA, 2015.
- GraphPad Prism; Version 8.4.2; GraphPad: San Diego, CA, USA, 2019.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Score | Clinical Manifestation |
|---|---|
| 0 | Absence of symptoms defined by the ability of the mouse to fully extend its hind limbs away from the body midline at least for 2 s during the tail suspension test (repeated 2–3 times). |
| 1 | The mouse shows a collapse of its hind limbs extension away from the body midline or a trembling of the hind limbs during tail suspension, indicating a beginning of hind limb paresis. |
| 2 | The mouse is dragging along any part of the feet or curling the toes during walking. |
| 3 | Signs of rigid paresis appear along with the feet not being used for forward motion. |
| 4 | The mouse is not able to right itself within 30 s from either side. Therewith, the human endpoint is reached. |
| 1st Antibody | 2nd Antibody | Final Dilution 1st Antibody | Final Dilution 2nd Antibody | Volume (per Sample) |
|---|---|---|---|---|
| Rabbit anti-βIII-tubulin RRID: AB_262133 | Alexa Fluor® 488-conjugated donkey anti-rabbit IgG (H + L) RRID: AB_2535792 | 1:3000 | 1:2000 | 250 µL |
| Rabbit anti-CA-II RRID: AB_2065996 | 1:250 | 1:2000 | 250 µL | |
| Rabbit anti-Iba-1 RRID: AB_839504 | 1:500 | 1:2000 | 250 µL | |
| Rabbit anti-S100βRRID: AB_2620024 | 1:5000 | 1:2000 | 250 µL | |
| Rat anti-CD68RRID: AB_324217 | Alexa Fluor® 488-conjugated goat anti-rat IgG (H + L) RRID: AB_2534074 | 1:100 | 1:2000 | 250 µL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ain, Q.; Schmeer, C.W.; Wengerodt, D.; Hofmann, Y.; Witte, O.W.; Kretz, A. Optimized Protocol for Proportionate CNS Cell Retrieval as a Versatile Platform for Cellular and Molecular Phenomapping in Aging and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 3000. https://doi.org/10.3390/ijms23063000
Ain Q, Schmeer CW, Wengerodt D, Hofmann Y, Witte OW, Kretz A. Optimized Protocol for Proportionate CNS Cell Retrieval as a Versatile Platform for Cellular and Molecular Phenomapping in Aging and Neurodegeneration. International Journal of Molecular Sciences. 2022; 23(6):3000. https://doi.org/10.3390/ijms23063000
Chicago/Turabian StyleAin, Quratul, Christian W. Schmeer, Diane Wengerodt, Yvonne Hofmann, Otto W. Witte, and Alexandra Kretz. 2022. "Optimized Protocol for Proportionate CNS Cell Retrieval as a Versatile Platform for Cellular and Molecular Phenomapping in Aging and Neurodegeneration" International Journal of Molecular Sciences 23, no. 6: 3000. https://doi.org/10.3390/ijms23063000
APA StyleAin, Q., Schmeer, C. W., Wengerodt, D., Hofmann, Y., Witte, O. W., & Kretz, A. (2022). Optimized Protocol for Proportionate CNS Cell Retrieval as a Versatile Platform for Cellular and Molecular Phenomapping in Aging and Neurodegeneration. International Journal of Molecular Sciences, 23(6), 3000. https://doi.org/10.3390/ijms23063000