
Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain



Abstract
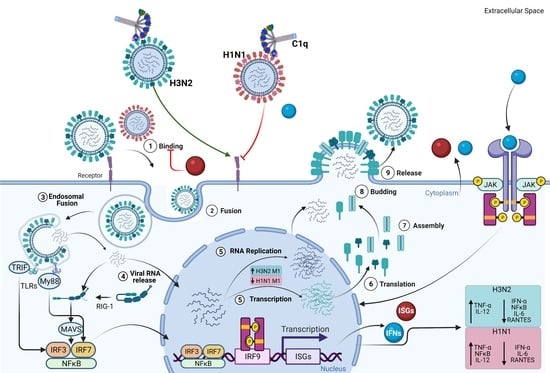
:
1. Introduction
2. Results
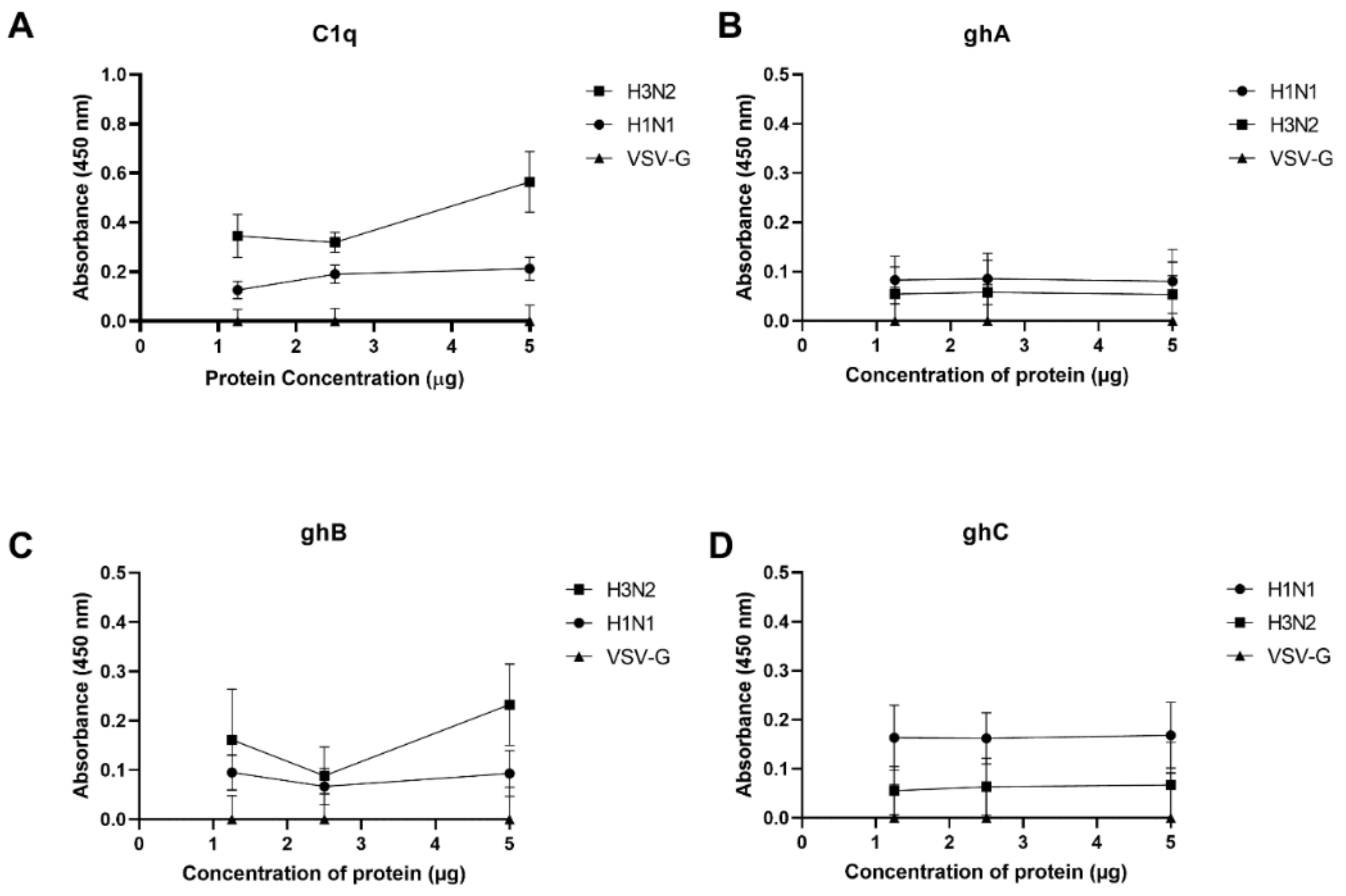
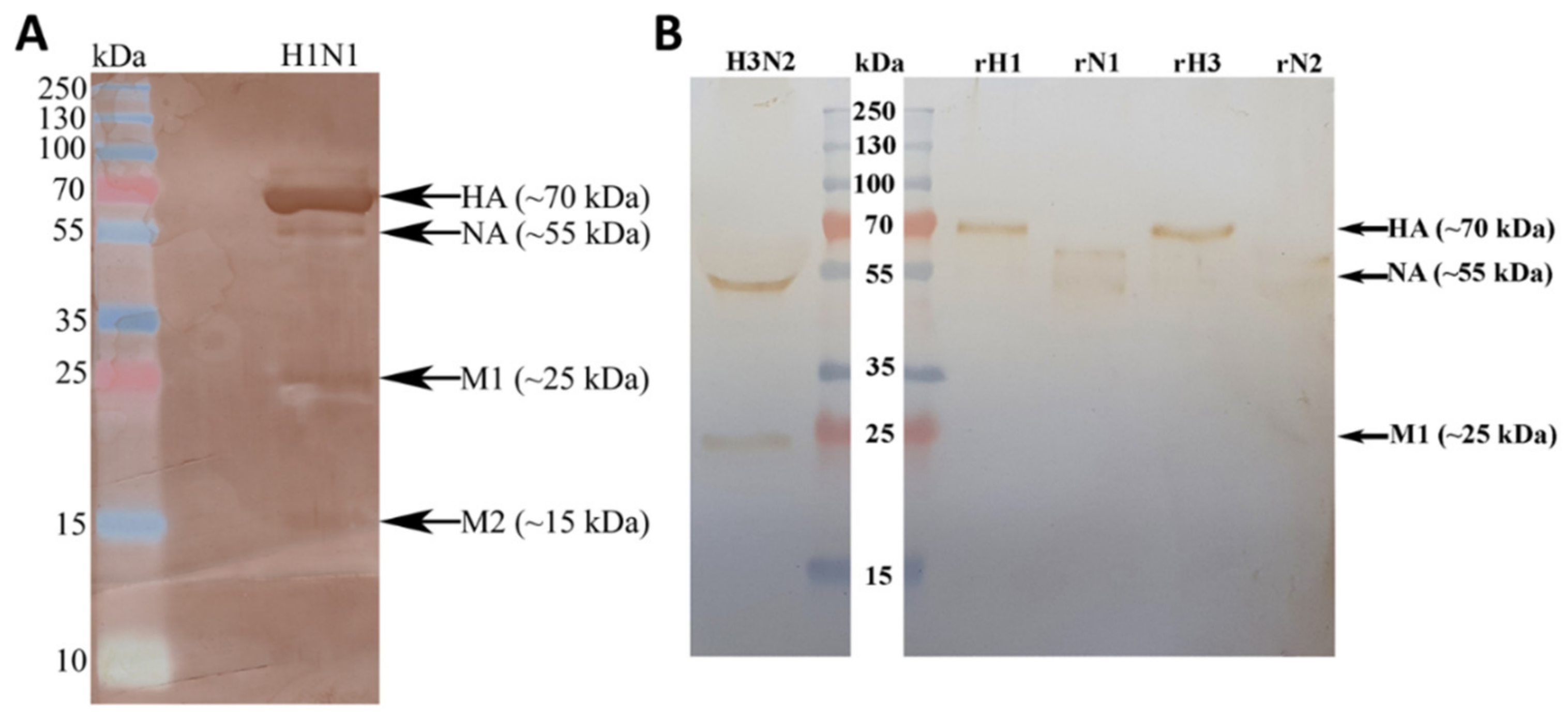
2.1. Immobilised Human C1q and Its Recombinant Globular Head Modules Bind Influenza A Virus Subtypes
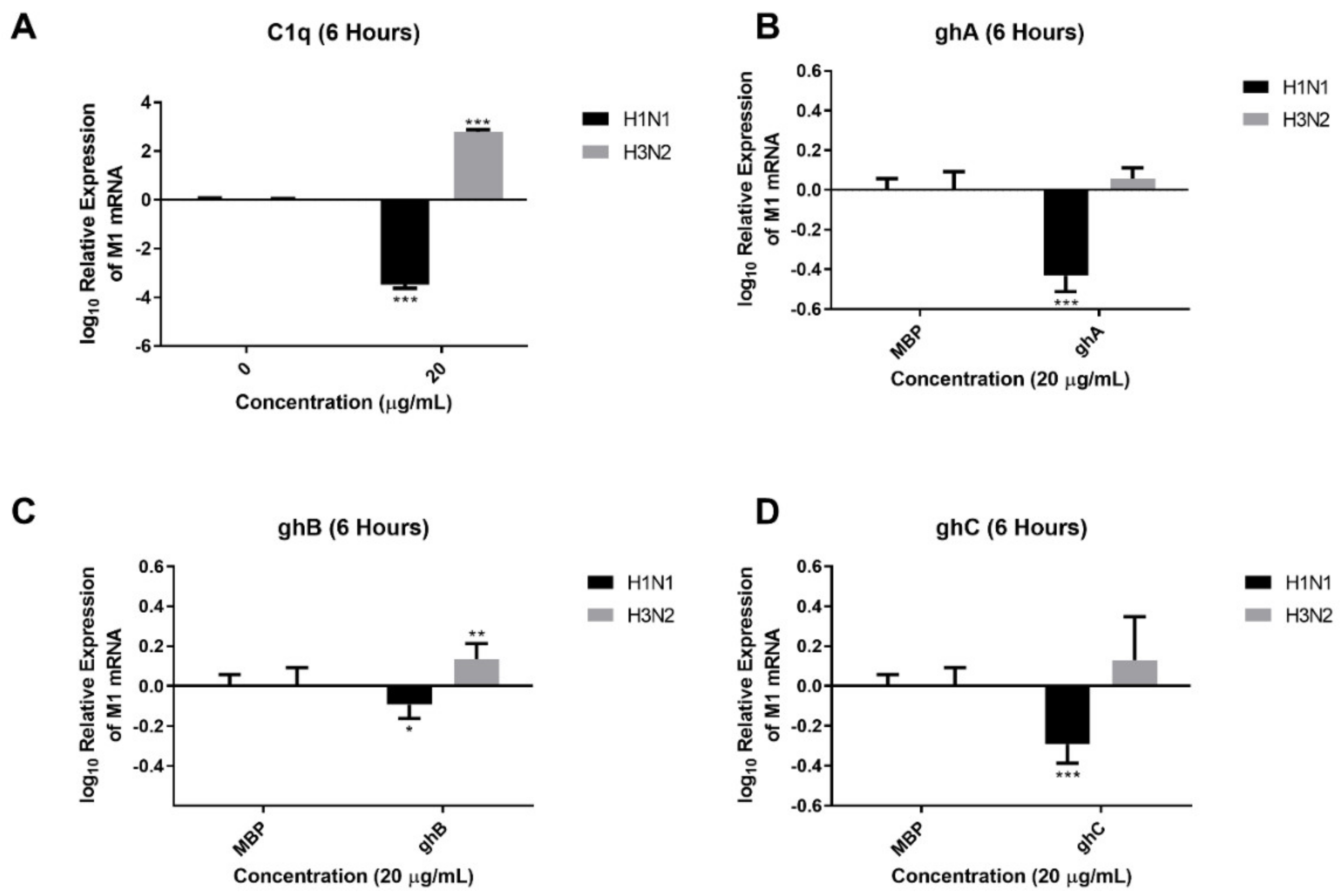
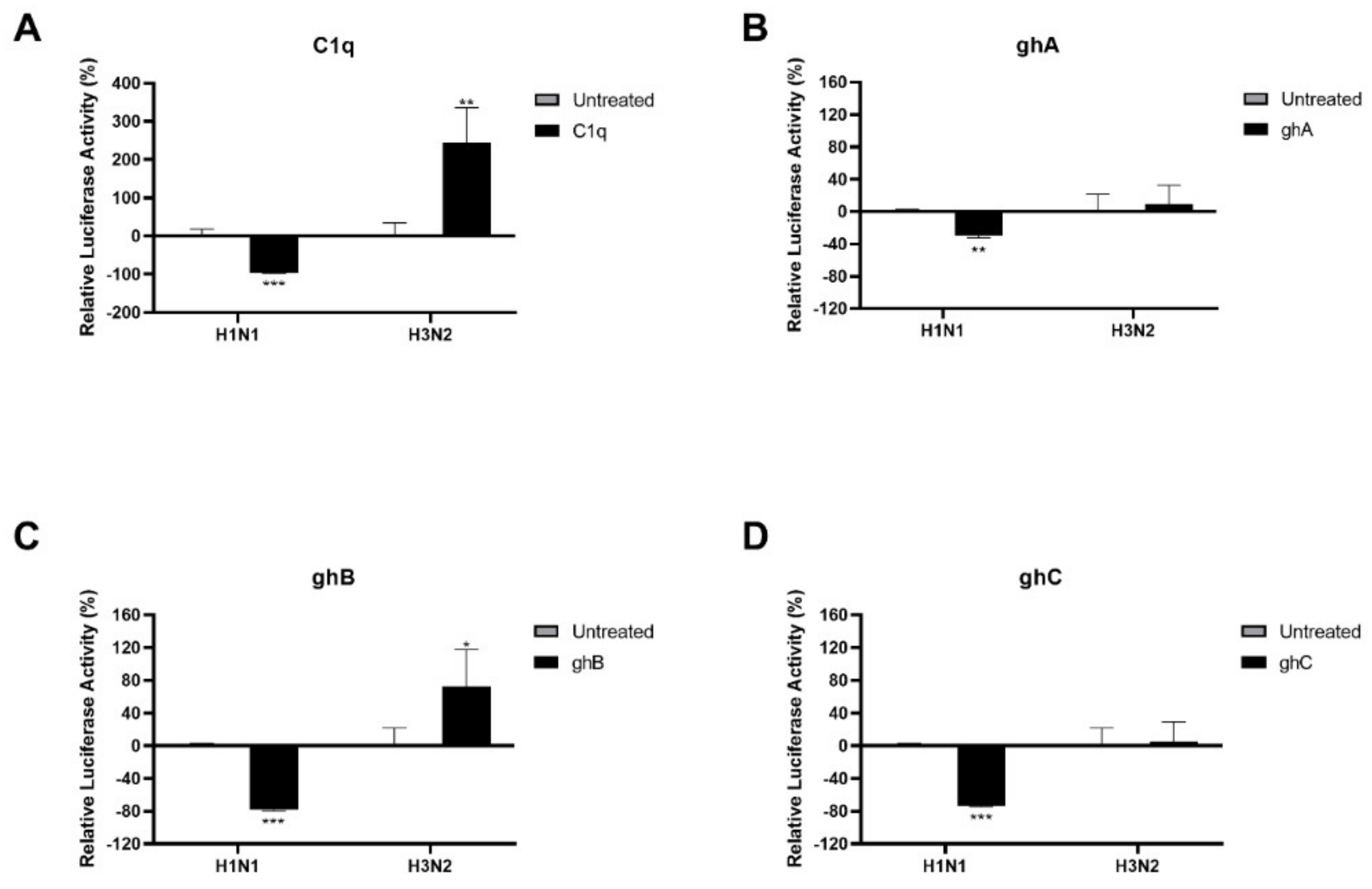
2.2. Human C1q Modulates IAV Replication in A549 Lung Epithelial Cells
2.3. Human C1q Modulates IAV Entry in A549 Lung Epithelial Cells by Inhibiting IAV-Cell Interaction
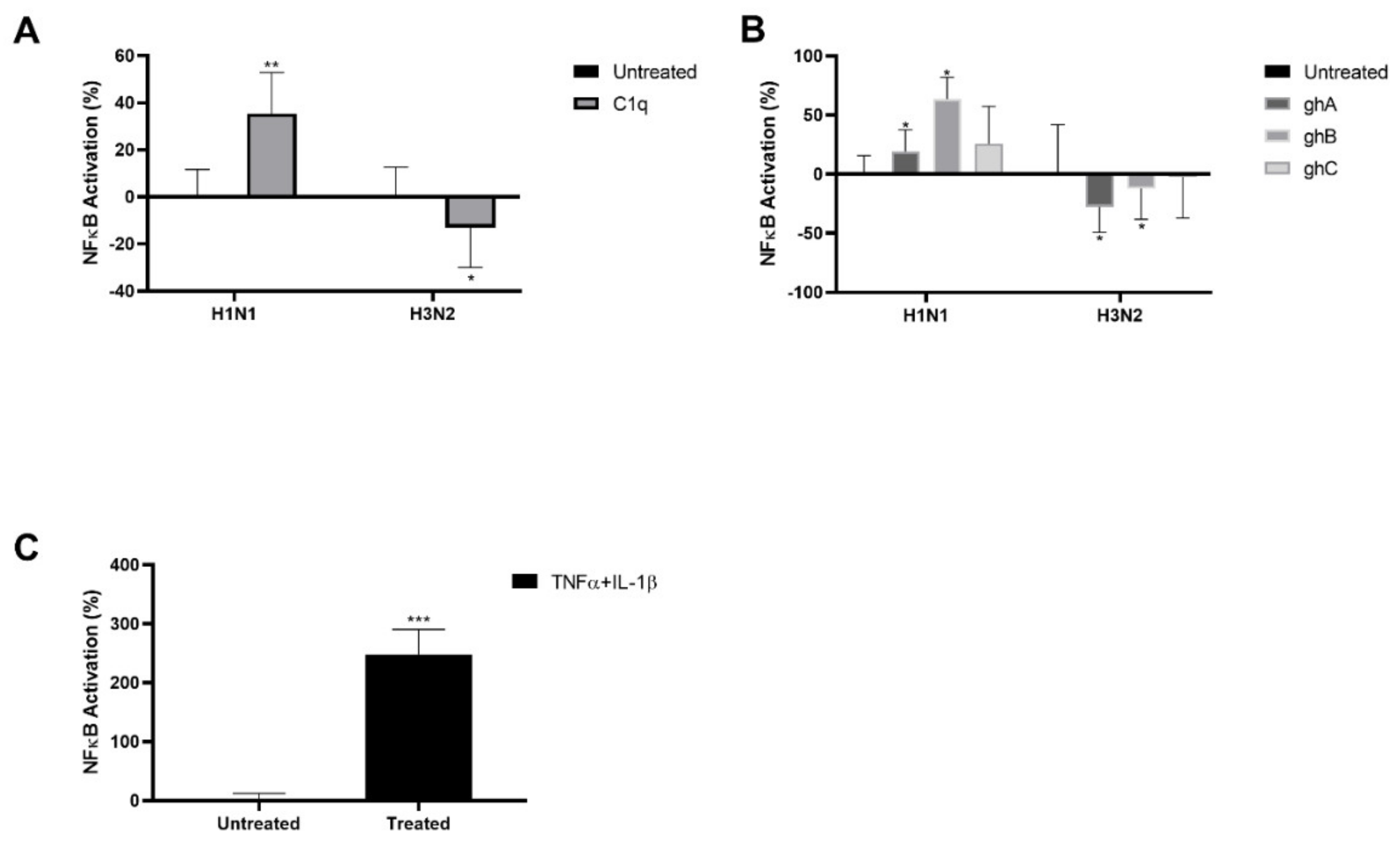
2.4. IAV Infection Induced NF–κB Activation Is Modulated by Human C1q
2.5. Modulation of Immune Responses by C1q on A549 Cells Infected with IAV Subtypes
3. Discussion
4. Materials and Methods
4.1. Purification of Human C1q
4.2. Production of Recombinant Globular Heads of Human C1q
4.3. Preparation of Viral Stocks and Titration
4.4. Direct Binding ELISA
4.5. Far-Western Blotting
4.6. Infection Assay and qRT-PCR Analysis
4.7. NF-κB Activation Assay
4.8. Production of Pseudo-Typed Lentiviral Particles
4.9. Entry Inhibition Assay
4.10. Cell Binding Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simonsen, L.; Viboud, C.; Taylor, R.J.; Miller, M.A. The Epidemiology of Influenza and Its Control. In Influenza Vaccines for the Future; Rappuoli, R., Del Giudice, G., Eds.; Springer: Basel, Switzerland, 2011; pp. 27–54. [Google Scholar]
- Bresee, J.S.; Fry, A.M.; Sambhara, S.; Cox, N.J. Inactivated Influenza Vaccines. In Plotkin’s Vaccines, 7th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Edwards, K.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 456–488.e21. [Google Scholar]
- Taubenberger, J.K.; Morens, D.M. The Pathology of Influenza Virus Infections. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 499–522. [Google Scholar] [CrossRef]
- García-Sastre, A. Influenza. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 414–420. [Google Scholar]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [Green Version]
- CDC The 2009 H1N1 Pandemic: Summary Highlights, April 2009–April 2010. Available online: https://www.cdc.gov/h1n1flu/cdcresponse.htm (accessed on 22 November 2021).
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153. [Google Scholar] [PubMed]
- Leung, H.S.; Li, O.T.; Chan, R.W.; Chan, M.C.; Nicholls, J.M.; Poon, L.L. Entry of influenza A Virus with a alpha2,6-linked sialic acid binding preference requires host fibronectin. J. Virol. 2012, 86, 10704–10713. [Google Scholar] [CrossRef] [Green Version]
- Steinhauer, D.; Wharton, S. Structure and Function of the Haemagglutinin. In Textbook of Influenza; Nicholson, K.G., Webster, R.G., Hay, A.J., Eds.; Blackwell Science: London, UK, 1998; Volume 1, pp. 54–64. [Google Scholar]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor Binding and Membrane Fusion in Virus Entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Wilson, I.A.; Skehel, J.J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981, 289, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Shtyrya, Y.A.; Mochalova, L.V.; Bovin, N.V. Influenza virus neuraminidase: Structure and function. Acta Nat. 2009, 1, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.-D. Neuraminidase Is Important for the Initiation of Influenza Virus Infection in Human Airway Epithelium. J. Virol. 2004, 78, 12665–12667. [Google Scholar] [CrossRef] [Green Version]
- Holsinger, L.J.; Alams, R. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology 1991, 183, 32–43. [Google Scholar] [CrossRef]
- Martin, K.; Helenius, A. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 1991, 65, 232–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieczkarski, S.B.; Whittaker, G.R. Viral Entry. In Membrane Trafficking in Viral Replication; Marsh, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 1–23. [Google Scholar]
- Stegmann, T. Membrane Fusion Mechanisms: The Influenza Hemagglutinin Paradigm and its Implications for Intracellular Fusion. Traffic 2000, 1, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F.; et al. NRAV, a Long Noncoding RNA, Modulates Antiviral Responses through Suppression of Interferon-Stimulated Gene Transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Munir, M. TRIM Proteins: Another Class of Viral Victims. Sci. Signal. 2010, 3, jc2. [Google Scholar] [CrossRef]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef]
- Hiscott, J.; Lin, R.; Nakhaei, P.; Paz, S. MasterCARD: A priceless link to innate immunity. Trends Mol. Med. 2006, 12, 53–56. [Google Scholar] [CrossRef]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.-T.; Flavell, R.A.; Liljeström, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Poeck, H.; Anz, D.; Berger, M.; Schlee, M.; Kim, S.; Bourquin, C.; Goutagny, N.; Jiang, Z.; Fitzgerald, K.A.; et al. Selection of Molecular Structure and Delivery of RNA Oligonucleotides to Activate TLR7 versus TLR8 and to Induce High Amounts of IL-12p70 in Primary Human Monocytes. J. Immunol. 2009, 182, 6824–6833. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.Y.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN Triggers RIG-I/TLR3/NLRP3-dependent Inflammasome Activation in Influenza A Virus Infected Cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef] [Green Version]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential Expression of NLRP3 among Hematopoietic Cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Mayor, A.; Tschopp, J. The Inflammasomes: Guardians of the Body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 Inflammasome by IAV Virulence Protein PB1-F2 Contributes to Severe Pathophysiology and Disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Kumar, P.; Malarkannan, S. Evasion of natural killer cells by influenza virus. J. Leukoc. Biol. 2011, 89, 189–194. [Google Scholar] [CrossRef] [Green Version]
- van Helden, M.J.G.; de Graaf, N.; Boog, C.J.P.; Topham, D.J.; Zaiss, D.M.W.; Sijts, A.J.A.M. The Bone Marrow Functions as the Central Site of Proliferation for Long-Lived NK Cells. J. Immunol. 2012, 189, 2333–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, M.; Tekoah, Y.; Zilka, A.; Gershoni-Yahalom, O.; Gazit, R.; Achdout, H.; Bovin, N.V.; Meningher, T.; Mandelboim, M.; Mandelboim, O.; et al. NKp46 O-Glycan Sequences That Are Involved in the Interaction with Hemagglutinin Type 1 of Influenza Virus. J. Virol. 2010, 84, 3789–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heer, A.K.; Harris, N.L.; Kopf, M.; Marsland, B.J. CD4+and CD8+ T Cells Exhibit Differential Requirements for CCR7-Mediated Antigen Transport during Influenza Infection. J. Immunol. 2008, 181, 6984–6994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintzen, G.; Ohl, L.; del Rio, M.-L.; Rodriguez-Barbosa, J.-I.; Pabst, O.; Kocks, J.R.; Krege, J.; Hardtke, S.; Förster, R. Induction of Tolerance to Innocuous Inhaled Antigen Relies on a CCR7-Dependent Dendritic Cell-Mediated Antigen Transport to the Bronchial Lymph Node. J. Immunol. 2006, 177, 7346–7354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GeurtsvanKessel, C.H.; Willart, M.A.M.; van Rijt, L.S.; Muskens, F.; Kool, M.; Baas, C.; Thielemans, K.; Bennett, C.; Clausen, B.E.; Hoogsteden, H.C.; et al. Clearance of influenza virus from the lung depends on migratory langerin +CD11b− but not plasmacytoid dendritic cells. J. Exp. Med. 2008, 205, 1621–1634. [Google Scholar] [CrossRef] [Green Version]
- Van de Sandt, C.E.; Kreijtz, J.H.C.M.; Rimmelzwaan, G.F. Evasion of Influenza A Viruses from Innate and Adaptive Immune Responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef] [Green Version]
- GeurtsvanKessel, C.H.; Bergen, I.M.; Muskens, F.; Boon, L.; Hoogsteden, H.C.; Osterhaus, A.D.M.E.; Rimmelzwaan, G.F.; Lambrecht, B.N. Both Conventional and Interferon Killer Dendritic Cells Have Antigen-Presenting Capacity during Influenza Virus Infection. PLoS ONE 2009, 4, e7187. [Google Scholar] [CrossRef] [Green Version]
- Tumpey, T.M.; García-Sastre, A.; Taubenberger, J.K.; Palese, P.; Swayne, D.E.; Pantin-Jackwood, M.J.; Schultz-Cherry, S.; Solórzano, A.; Rooijen, N.V.; Katz, J.M.; et al. Pathogenicity of Influenza Viruses with Genes from the 1918 Pandemic Virus: Functional Roles of Alveolar Macrophages and Neutrophils in Limiting Virus Replication and Mortality in Mice. J. Virol. 2005, 79, 14933–14944. [Google Scholar] [CrossRef] [Green Version]
- Murugaiah, V.; Varghese, P.M.; Saleh, S.M.; Tsolaki, A.G.; Alrokayan, S.H.; Khan, H.A.; Collison, K.S.; Sim, R.B.; Nal, B.; Al-Mohanna, F.A.; et al. Complement-Independent Modulation of Influenza A Virus Infection by Factor H. Front. Immunol. 2020, 11, 355. [Google Scholar] [CrossRef]
- Sim, R.B.; Tsiftsoglou, S.A. Proteases of the complement system. Biochem. Soc. Trans. 2004, 32, 21–27. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Nandakumar, K.S.; Tedesco, F. Inhibiting the C5–C5a receptor axis. Mol. Immunol. 2011, 48, 1631–1642. [Google Scholar] [CrossRef]
- Muller-Eberhard, H.J. The Membrane Attack Complex of Complement. Annu. Rev. Immunol. 1986, 4, 503–528. [Google Scholar] [CrossRef]
- Kopf, M.; Abel, B.; Gallimore, A.; Carroll, M.; Bachmann, M.F. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat. Med. 2002, 8, 373–378. [Google Scholar] [CrossRef]
- Hicks, J.T.; Ennis, F.A.; Kim, E.; Verbonitz, M. The Importance of an Intact Complement Pathway in Recovery from a Primary Viral Infection: Influenza in Decomplemented and in C5-Deficient Mice. J. Immunol. 1978, 121, 1437–1445. [Google Scholar]
- Kandasamy, M.; Ying, P.C.; Ho, A.W.S.; Sumatoh, H.R.; Schlitzer, A.; Hughes, T.R.; Kemeny, D.M.; Morgan, B.P.; Ginhoux, F.; Sivasankar, B. Complement Mediated Signaling on Pulmonary CD103+ Dendritic Cells Is Critical for Their Migratory Function in Response to Influenza Infection. PLoS Pathog. 2013, 9, e1003115. [Google Scholar] [CrossRef] [Green Version]
- Carroll, M.C.; Isenman, D.E. Regulation of Humoral Immunity by Complement. Immunity 2012, 37, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Jayasekera, J.P.; Moseman, E.A.; Carroll, M.C. Natural Antibody and Complement Mediate Neutralization of Influenza Virus in the Absence of Prior Immunity. J. Virol. 2007, 81, 3487–3494. [Google Scholar] [CrossRef] [Green Version]
- Rattan, A.; Pawar, S.D.; Nawadkar, R.; Kulkarni, N.; Lal, G.; Mullick, J.; Sahu, A. Synergy between the classical and alternative pathways of complement is essential for conferring effective protection against the pandemic influenza A(H1N1) 2009 virus infection. PLoS Pathog. 2017, 13, e1006248. [Google Scholar] [CrossRef] [Green Version]
- Mozdzanowska, K.; Feng, J.; Eid, M.; Zharikova, D.; Gerhard, W. Enhancement of neutralizing activity of influenza virus-specific antibodies by serum components. Virology 2006, 352, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.Q.; Mozdzanowska, K.; Gerhard, W. Complement component C1q enhances the biological activity of influenza virus hemagglutinin-specific antibodies depending on their fine antigen specificity and heavy-chain isotype. J. Virol. 2002, 76, 1369–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, G.; Liu, X.; Wang, Z.; Liu, W.; Ye, X. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J. Gen. Virol. 2009, 90, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Castellano, G.; Woltman, A.M.; Nauta, A.J.; Roos, A.; Trouw, L.A.; Seelen, M.A.; Schena, F.P.; Daha, M.R.; van Kooten, C. Maturation of dendritic cells abrogates C1q production In Vivo and In Vitro. Blood 2004, 103, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Day, N.K.; Gewurz, H.; Pickering, R.J.; Good, R.A. Ontogenetic Development of Clq Synthesis in the Piglet. J. Immunol. 1970, 104, 1316–1319. [Google Scholar]
- Kaul, M.; Loos, M. Expression of membrane C1q in human monocyte-derived macrophages is developmentally regulated and enhanced by interferon-γ. FEBS Lett. 2001, 500, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Petry, F.; Botto, M.; Holtappels, R.; Walport, M.J.; Loos, M. Reconstitution of the Complement Function in C1q-Deficient (C1qa−/−) Mice with Wild-Type Bone Marrow Cells. J. Immunol. 2001, 167, 4033–4037. [Google Scholar] [CrossRef] [Green Version]
- Stecher, V.J.; Morse, J.H.; Thorbecke, G.J. Sites of Production of Primate Serum Proteins Associated with the Complement System. Proc. Soc. Exp. Biol. Med. 1967, 124, 433–438. [Google Scholar] [CrossRef]
- Kishore, U.; Gaboriaud, C.; Waters, P.; Shrive, A.K.; Greenhough, T.J.; Reid, K.B.M.; Sim, R.B.; Arlaud, G.J. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol. 2004, 25, 551–561. [Google Scholar] [CrossRef]
- Kishore, U.; Reid, K.B.M. C1q: Structure, function, and receptors. Immunopharmacology 2000, 49, 159–170. [Google Scholar] [CrossRef]
- Thielens, N.M.; Tacnet-Delorme, P.; Arlaud, G.J. Interaction of C1q and Mannan-Binding Lectin with Viruses. Immunobiology 2002, 205, 563–574. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Vieillard, V.; Sagan, S.; Bismuth, G.; Debré, P. HIV gp41 Engages gC1qR on CD4+ T Cells to Induce the Expression of an NK Ligand through the PIP3/H2O2 Pathway. PLoS Pathog. 2010, 6, e1000975. [Google Scholar] [CrossRef]
- Ebenbichler, C.F.; Thielens, N.M.; Vornhagen, R.; Marschang, P.; Arlaud, G.J.; Dierich, M.P. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J. Exp. Med. 1991, 174, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Thielens, N.M.; Bally, I.M.; Ebenbichler, C.F.; Dierich, M.P.; Arlaud, G.J. Further characterization of the interaction between the C1q subcomponent of human C1 and the transmembrane envelope glycoprotein gp41 of HIV-1. J. Immunol. 1993, 151, 6583–6592. [Google Scholar]
- Kishore, U.; Gupta, S.K.; Perdikoulis, M.V.; Kojouharova, M.S.; Urban, B.C.; Reid, K.B.M. Modular Organization of the Carboxyl-Terminal, Globular Head Region of Human C1q A, B, and C Chains. J. Immunol. 2003, 171, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Kittlesen, D.J.; Chianese-Bullock, K.A.; Yao, Z.Q.; Braciale, T.J.; Hahn, Y.S. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J. Clin. Investig. 2000, 106, 1239–1249. [Google Scholar] [CrossRef]
- Mohan, K.V.K.; Ghebrehiwet, B.; Atreya, C.D. The N-terminal conserved domain of rubella virus capsid interacts with the C-terminal region of cellular p32 and overexpression of p32 enhances the viral infectivity. Virus Res. 2002, 85, 151–161. [Google Scholar] [CrossRef]
- Matthews, D.A.; Russell, W.C. Adenovirus core protein V interacts with p32--a protein which is associated with both the mitochondria and the nucleus. J. Gen. Virol. 1998, 79, 1677–1685. [Google Scholar] [CrossRef]
- Wang, Y.; Finan, J.E.; Middeldorp, J.M.; Hayward, S.D. P32/TAP, a Cellular Protein That Interacts with EBNA-1 of Epstein–Barr Virus. Virology 1997, 236, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Kunnakkadan, U.; Nag, J.; Kumar, N.A.; Mukesh, R.K.; Suma, S.M.; Johnson, J.B.; Dutch, R.E. Complement-Mediated Neutralization of a Potent Neurotropic Human Pathogen, Chandipura Virus, Is Dependent on C1q. J. Virol. 2019, 93, e00994-19. [Google Scholar] [CrossRef]
- Varghese, P.M.; Murugaiah, V.; Beirag, N.; Temperton, N.; Khan, H.A.; Alrokayan, S.H.; Al-Ahdal, M.N.; Nal, B.; Al-Mohanna, F.A.; Sim, R.B.; et al. C4b Binding Protein Acts as an Innate Immune Effector Against Influenza A Virus. Front. Immunol. 2021, 11, 3356. [Google Scholar] [CrossRef] [PubMed]
- La Gruta, N.L.; Kedzierska, K.; Stambas, J.; Doherty, P.C. A question of self-preservation: Immunopathology in influenza virus infection. Immunol. Cell Biol. 2007, 85, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Shinya, K.; Gao, Y.; Cilloniz, C.; Suzuki, Y.; Fujie, M.; Deng, G.; Zhu, Q.; Fan, S.; Makino, A.; Muramoto, Y.; et al. Integrated Clinical, Pathologic, Virologic, and Transcriptomic Analysis of H5N1 Influenza Virus-Induced Viral Pneumonia in the Rhesus Macaque. J. Virol. 2012, 86, 6055–6066. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Planz, O. Influenza viruses and the NF-kappaB signaling pathway—Towards a novel concept of antiviral therapy. Biol. Chem. 2008, 389, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Oslund, K.L.; Baumgarth, N. Influenza-induced innate immunity: Regulators of viral replication, respiratory tract pathology & adaptive immunity. Future Virol. 2011, 6, 951–962. [Google Scholar]
- Lee, S.M.; Cheung, C.Y.; Nicholls, J.M.; Hui, K.P.; Leung, C.Y.; Uiprasertkul, M.; Tipoe, G.L.; Lau, Y.L.; Poon, L.L.; Ip, N.Y.; et al. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: A mechanism for the pathogenesis of avian influenza H5N1 infection. J. Infect. Dis. 2008, 198, 525–535. [Google Scholar] [CrossRef]
- Yarilina, A.; Park-Min, K.H.; Antoniv, T.; Hu, X.; Ivashkiv, L.B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 2008, 9, 378–387. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Al-Ahdal, M.N.; Murugaiah, V.; Varghese, P.M.; Abozaid, S.M.; Saba, I.; Al-Qahtani, A.A.; Pathan, A.A.; Kouser, L.; Nal, B.; Kishore, U. Entry Inhibition and Modulation of Pro-Inflammatory Immune Response Against Influenza A Virus by a Recombinant Truncated Surfactant Protein D. Front. Immunol. 2018, 9, 1586. [Google Scholar] [CrossRef]
- Kim, K.S.; Jung, H.; Shin, I.K.; Choi, B.-R.; Kim, D.H. Induction of interleukin-1 beta (IL-1β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J. Med. Virol. 2015, 87, 1104–1112. [Google Scholar] [CrossRef]
- Ramos, I.; Bernal-Rubio, D.; Durham, N.; Belicha-Villanueva, A.; Lowen, A.C.; Steel, J.; Fernandez-Sesma, A. Effects of Receptor Binding Specificity of Avian Influenza Virus on the Human Innate Immune Response. J. Virol. 2011, 85, 4421–4431. [Google Scholar] [CrossRef] [Green Version]
- Ramos, I.; Fernandez-Sesma, A. Modulating the Innate Immune Response to Influenza A Virus: Potential Therapeutic Use of Anti-Inflammatory Drugs. Front. Immunol. 2015, 6, 361. [Google Scholar] [CrossRef] [Green Version]
- Muramoto, Y.; Shoemaker, J.E.; Le, M.Q.; Itoh, Y.; Tamura, D.; Sakai-Tagawa, Y.; Imai, H.; Uraki, R.; Takano, R.; Kawakami, E.; et al. Disease Severity Is Associated with Differential Gene Expression at the Early and Late Phases of Infection in Nonhuman Primates Infected with Different H5N1 Highly Pathogenic Avian Influenza Viruses. J. Virol. 2014, 88, 8981–8997. [Google Scholar] [CrossRef] [Green Version]
- Cilloniz, C.; Pantin-Jackwood, M.J.; Ni, C.; Goodman, A.G.; Peng, X.; Proll, S.C.; Carter, V.S.; Rosenzweig, E.R.; Szretter, K.J.; Katz, J.M.; et al. Lethal Dissemination of H5N1 Influenza Virus Is Associated with Dysregulation of Inflammation and Lipoxin Signaling in a Mouse Model of Infection. J. Virol. 2010, 84, 7613–7624. [Google Scholar] [CrossRef] [Green Version]
- Peiris, J.S.M.; Cheung, C.Y.; Leung, C.Y.H.; Nicholls, J.M. Innate immune responses to influenza A H5N1: Friend or foe? Trends Immunol. 2009, 30, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Duvigneau, S.; Sharma-Chawla, N.; Boianelli, A. Hierarchical effects of pro-inflammatory cytokines on the post-influenza susceptibility to pneumococcal coinfection. Sci. Rep. 2016, 6, 37045. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.-W.; Lu, J.; Wu, C.-L.; Yi, L.-N.; Xie, X.; Shi, X.-D.; Fang, S.-S.; Zan, H.; Kung, H.-f.; He, M.-L. Three fatal cases of pandemic 2009 influenza A virus infection in Shenzhen are associated with cytokine storm. Respir. Physiol. Neurobiol. 2011, 175, 185–187. [Google Scholar] [CrossRef]
- Dawson, T.C.; Beck, M.A.; Kuziel, W.A.; Henderson, F.; Maeda, N. Contrasting Effects of CCR5 and CCR2 Deficiency in the Pulmonary Inflammatory Response to Influenza A Virus. Am. J. Pathol. 2000, 156, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, A.; Salentin, R.; Meyer, R.G.; Bussfeld, D.; Pauligk, C.; Fesq, H.; Hofmann, P.; Nain, M.; Gemsa, D.; Sprenger, H. Defense against Influenza A Virus Infection: Essential Role of the Chemokine System. Immunobiology 2001, 204, 603–613. [Google Scholar] [CrossRef]
- Tan, L.A.; Yu, B.; Sim, F.C.J.; Kishore, U.; Sim, R.B. Complement activation by phospholipids: The interplay of factor H and C1q. Protein Cell 2010, 1, 1033–1049. [Google Scholar] [CrossRef] [Green Version]
- Klimov, A.; Balish, A.; Veguilla, V.; Sun, H.; Schiffer, J.; Lu, X.; Katz, J.M.; Hancock, K. Influenza Virus Titration, Antigenic Characterization, and Serological Methods for Antibody Detection. In Influenza Virus: Methods and Protocols; Kawaoka, Y., Neumann, G., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 25–51. [Google Scholar]
- Tang, D.-J.; Lam, Y.-M.; Siu, Y.-L.; Lam, C.-H.; Chu, S.-L.; Peiris, J.S.M.; Buchy, P.; Nal, B.; Bruzzone, R. A Single Residue Substitution in the Receptor-Binding Domain of H5N1 Hemagglutinin Is Critical for Packaging into Pseudotyped Lentiviral Particles. PLoS ONE 2012, 7, e43596. [Google Scholar] [CrossRef] [Green Version]
- Varghese, P.M.; Mukherjee, S.; Al-Mohanna, F.A.; Saleh, S.M.; Almajhdi, F.N.; Beirag, N.; Alkahtani, S.H.; Rajkumari, R.; Rogier, B.N.; Sim, R.B.; et al. Human Properdin Released by Infiltrating Neutrophils Can Modulate Influenza A Virus Infection. Front. Immunol. 2021, 12, 747654. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | C1q | ghA | ghB | ghC | ||||
| 2 h | 6 h | 2 h | 6 h | 2 h | 6 h | 2 h | 6 h | |
| TNF-α | − | ↑ (0.15 log10) | ↑ (2.4 log10) | ↑ (0.63 log10) | ↑ (3.20 log10) | ↑ (0.27 log10) | ↑ (2.82 log10) | ↑ (0.03 log10) |
| NFkB | − | ↑ (0.09 log10) | ↑ (0.018 log10) | ↑ (0.14 log10) | ↑ (0.33 log10) | ↑ (0.15 log10) | ↑ (0.47 log10) | ↑ (0.04 log10) |
| IFN-α | − | ↓ (−0.52 log10) | − | ↑ (0.06 log10) | ↓ (0.45 log10) | ↓ (0.10 log10) | ↓ (−0.25 log10) | ↓ (−0.55 log10) |
| IL-6 | − | ↓ (−0.29 log10) | − | ↑ (0.50 log10) | ↑ (1.98 log10) | ↑ (0.18 log10) | ↑ (1.84 log10) | ↓ (−0.05 log10) |
| IL-12 | − | ↑ (0.51 log10) | ↓ (−0.93 log10) | ↑ (1.17 log10) | ↓ (−0.14 log10) | ↑ (0.34 log10) | ↑ (0.19 log10) | ↑ (0.23 log10) |
| RANTES | − | ↓ (−0.018 log10) | ↓ (−0.39 log10) | ↑ (0.17 log10) | ↓ (−0.40 log10) | ↑ (0.23 log10) | ↑ (0.04 log10) | ↓ (−0.52 log10) |
| B | C1q | ghA | ghB | ghC | ||||
| 2 h | 6 h | 2 h | 6 h | 2 h | 6 h | 2 h | 6 h | |
| TNF-α | ↓ (−0.32 log10) | ↑ (0.37 log10) | ↓ (−0.29 log10) | ↑ (0.04 log10) | ↑ (0.31 log10) | ↓ (−0.25 log10) | ↓ (−0.11 log10) | ↑ (0.39 log10) |
| NF-κB | ↓ (−0.15 log10) | ↓ (−0.20 log10) | ↑ (0.77 log10) | ↓ (−0.42 log10) | ↑ (0.60 log10) | ↓ (−0.21 log10) | ↑ (0.65 log10) | ↓ (−0.04 log10) |
| IFN-α | ↓ (−2.12 log10) | ↓ (−0.07 log10) | ↓ (−0.96 log10) | ↓ (−1.88 log10) | ↓ (−2.15 log10) | ↓ (−0.23 log10) | ↓ (−0.51 log10) | ↓ (−1.75 log10) |
| IL-6 | ↑ (0.06 log10) | ↓ (−0.32 log10) | ↑ (0.30 log10) | ↓ (−0.83 log10) | ↑ (0.29 log10) | ↓ (−0.69 log10) | ↓ (−0.08 log10) | ↑ (0.25 log10) |
| IL-12 | − | ↑ (1.5 log10) | − | ↓ (−0.39 log10) | − | ↑ (0.71 log10) | − | ↓ (−0.24 log10) |
| RANTES | ↓ (−3.20 log10) | ↓ (−0.01 log10) | ↑ (−1.4 log10) | ↓ (−0.74 log10) | ↓ (−0.89 log10) | ↓ (−0.29 log10) | ↑ (0.35 log10) | ↓ (−1.36 log10) |
| Target | Forward Primer | Reverse Primer |
|---|---|---|
| 18S | 5′-ATGGCCGTTC TTAGTTGGTG-3′ | 5′-CGCTGAGCCA GTCAGTGTAG-3′ |
| IL-6 | 5′-GAAAGCAGCA AAGAGGCACT-3′ | 5′-TTTCACCAGG CAAGTCTCCT-3′ |
| IL-12 | 5′-AACTTGCAGC TGAAGCCATT-3′ | 5′-GACCTGAACG CAGAATGTCA-3′ |
| TNF-α | 5′-AGCCCATGTT GTAGCAAACC-3′ | 5′-TGAGGTACAG GCCCTCTGAT-3′ |
| M1 | 5′AAACATATGTCTGATAAC GAAGGAGAACAGTTCTT-3′ | 5′GCTGAATTCTACCT CATGGTCTTCTTGA-3′ |
| RANTES | 5′-GCGGGTACCAT GAAGATCTCTG-3′ | 5′-GGGTCAGAATC AAGAAACCCTC-3′ |
| IFN-α | 5′-TTT CTC CTG CC T GAA GGA CAG-3′ | 5′-GCT CAT GAT TTC TGC TCT GAC A-3′ |
| H1N1 | H3N2 | VSV-G | |
|---|---|---|---|
| Envelope Protein-Coding Plasmid | pcDNA3.1-swineH1-flag (H1 from swine H1N1 A/California/04/09) (Codon optimized H1 (Genecust)) | pcDNA-H3 (H3 from A/Denmark/70/03/(H3N2)) (Codon optimized H3 (Geneart)) | pCMV-VSV-G (Addgene plasmid # 8454) |
| pcDNA3.1-swine N1-flag (N1 from swine H1N1 A/California/04/09) (Codon optimised N1 (Genecust)) | pI.18-N2 (N2 from human H3N2 A/Texas/50/2012) | ||
| Backbone Plasmid | pHIV–Luciferase (Addgene plasmid # 21375) | ||
| Packaging Plasmid | psPAX2 (Addgene plasmid # 12260) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, P.M.; Kishore, U.; Rajkumari, R. Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain. Int. J. Mol. Sci. 2022, 23, 3045. https://doi.org/10.3390/ijms23063045
Varghese PM, Kishore U, Rajkumari R. Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain. International Journal of Molecular Sciences. 2022; 23(6):3045. https://doi.org/10.3390/ijms23063045
Chicago/Turabian StyleVarghese, Praveen M., Uday Kishore, and Reena Rajkumari. 2022. "Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain" International Journal of Molecular Sciences 23, no. 6: 3045. https://doi.org/10.3390/ijms23063045
APA StyleVarghese, P. M., Kishore, U., & Rajkumari, R. (2022). Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain. International Journal of Molecular Sciences, 23(6), 3045. https://doi.org/10.3390/ijms23063045