
Needles in Haystacks: Understanding the Success of Selective Pairing of Nucleic Acids

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Free Energy for Duplex Formation
2.1.1. Perfect Match: Initiation and Nearest-Neighbor Canonical Base Pairs
2.1.2. Dangling Ends: External Mismatches
2.1.3. Internal Mismatches
2.1.4. Salt Correction
2.1.5. CG Contribution
2.2. Degeneracy of Equivalent Duplexes
2.3. Quantifying Selectivity
3. Results
3.1. PCR
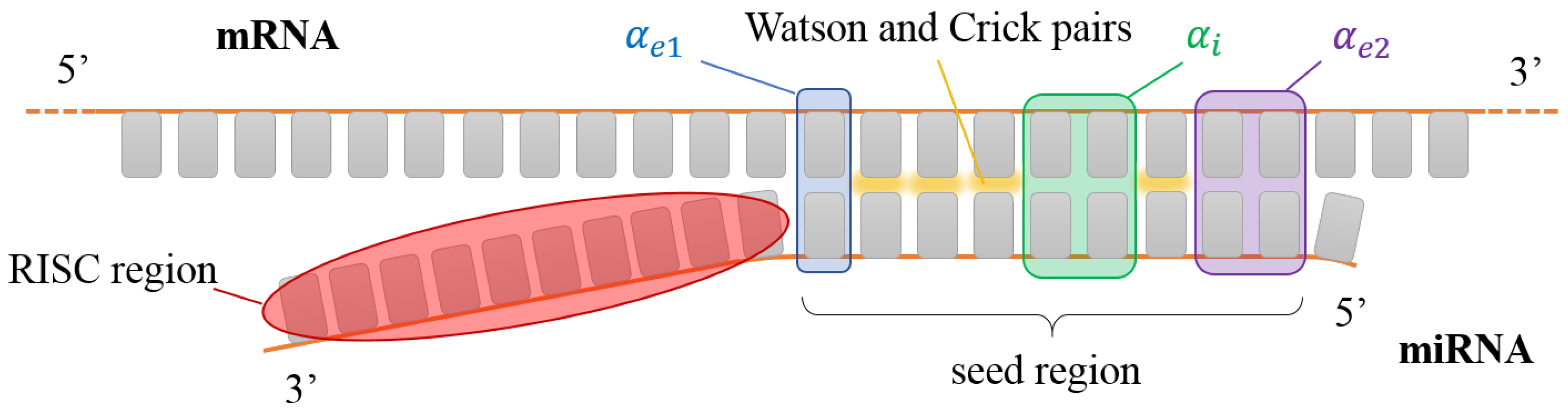
3.2. miRNA
4. Discussion
4.1. PCR
4.2. miRNA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Thermodynamic Parameters
Appendix A.1. Initiation and Canonical Watson–Crick Base Pairs
Appendix A.2. External and Internal Mismatches
Appendix A.3. Dependence on the Sequence, Fraction of CG
Appendix A.4. Salt Contribution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Propagation | Propagation | ||||
|---|---|---|---|---|---|
| Sequence | Sequence | ||||
| AA/TT | AA/UU | ||||
| AT/TA | AU/UA | ||||
| TA/AT | UA/AU | ||||
| CA/GT | CA/GU | ||||
| GT/CA | GU/CA | ||||
| CT/GA | CU/GA | ||||
| GA/CT | GA/CU | ||||
| CG/GC | CG/GC | ||||
| GC/CG | GC/CG | ||||
| GG/CC | GG/CC | ||||
| EC(G)/G(C)E | EC(G)/G(C)E | ||||
| EA(T)/T(A)E | EA(U)/U(A)E |
| X | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dangling | Propagation | A | T | C | G | ||||
| End | Sequence | ||||||||
| 5-dangling | XA/T | ||||||||
| XT/A | |||||||||
| XC/G | |||||||||
| XG/C | |||||||||
| 3-dangling | AX/T | ||||||||
| TX/A | |||||||||
| CX/G | |||||||||
| GX/C | |||||||||
| Y | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Propagation | A | T | C | G | |||||
| Sequence | X | ||||||||
| AX/TY | A | WC | WC | ||||||
| T | WC | WC | |||||||
| C | WC | WC | |||||||
| G | WC | WC | |||||||
| TX/AY | A | WC | WC | ||||||
| T | WC | WC | $0.7 | ||||||
| C | WC | WC | |||||||
| G | WC | WC | |||||||
| CX/GY | A | WC | WC | ||||||
| T | WC | WC | |||||||
| C | WC | WC | |||||||
| G | WC | WC | |||||||
| GX/CY | A | WC | WC | ||||||
| T | WC | WC | |||||||
| C | WC | WC | |||||||
| G | WC | WC | |||||||
| Propagation | ||
|---|---|---|
| Sequence | ||
| WW | ||
| SS | ||
| SW(WS) |
| L | |||||||
| °C | |||||||
| °C | |||||||
Appendix A.5. Estimation of the Goodness of the Thermodynamic Parameters

Appendix B. Melting Curve
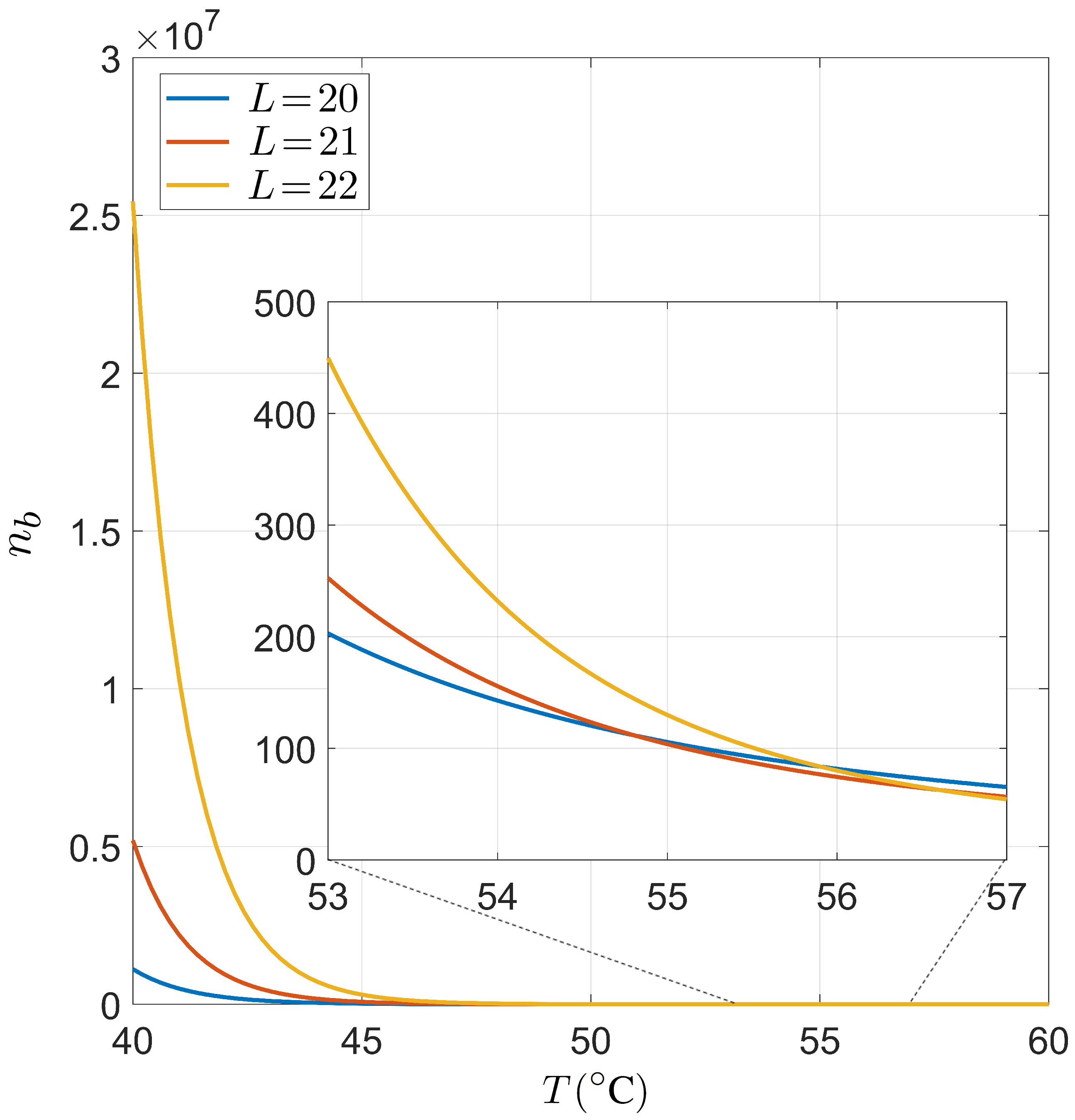
Appendix C. Role of the Salt Concentration

Appendix D. Disassociation Times
References
- Mullis, K.B.; Faloona, F.A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987, 155, 335–350. [Google Scholar] [PubMed]
- Van Pelt-Verkuil, E.; van Belkum, A.; Hays, J.P. PCR primers. In Principles and Technical Aspects of PCR Amplification; Springer: Dordrecht, The Netherlands, 2008; pp. 63–90. [Google Scholar]
- Chipman, L.B.; Pasquinelli, A.E. miRNA Targeting: Growing beyond the Seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirle, N.T.; Sheu-Gruttadauria, J.; MacRae, I.J. Structural Basis for microRNA Targeting. Science 2014, 346, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Lane, A.N.; Jenkins, T. Thermodynamics of nucleic acids and their interactions with ligands. Q. Rev. Biophys. 2000, 33, 255–306. [Google Scholar] [CrossRef]
- SantaLucia, J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 4, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Breslauer, K.J.; Frank, R.; Blöcker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [Google Scholar] [CrossRef] [Green Version]
- Owczarzy, R.; Vallone, P.M.; Gallo, F.J.; Paner, T.M.; Lane, M.J.; Benight, A.S. Predicting sequence-dependent melting stability of short duplex DNA oligomers. Biopolymers 1997, 44, 217–239. [Google Scholar] [CrossRef]
- Plata, C.A.; Marni, S.; Maritan, A.; Bellini, T.; Suweis, S. Statistical physics of DNA hybridization. Phys. Rev. E 2021, 103, 042503. [Google Scholar] [CrossRef]
- SantaLucia, J.; Hicks, D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. 2004, 33, 415–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vologodskii, A.; Frank-Kamenetskii, M.D. DNA melting and energetics of the double helix. Phys. Life Rev. 2018, 25, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Owczarzy, R.; You, Y.; Moreira, B.G.; Manthey, J.A.; Huang, L.; Behlke, M.A.; Walder, J.A. Effects of Sodium Ions on DNA Duplex Oligomers: Improved Predictions of Melting Temperatures. Biochemistry 2004, 43, 3537–3554. [Google Scholar] [CrossRef]
- Wu, J.H.; Hong, P.Y.; Liu, W.T. Quantitative effects of position and type of single mismatch on single base primer extension. J. Microbiol. Methods 2009, 77, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/ (accessed on 21 September 2020).
- Available online: https://www.proteinatlas.org/humanproteome/cell/cell+line (accessed on 21 September 2020).
- Kern, F.; Krammes, L.; Danz, K.; Diener, C.; Kehl, T.; Küchler, O.; Fehlmann, T.; Kahraman, M.; Rheinheimer, S.; Aparicio-Puerta, E.; et al. Validation of human microRNA target pathways enables evaluation of target prediction tools. Nucleic Acids Res. 2021, 49, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Tabunoki, H. Comprehensive analysis of human microRNA target networks. BioData Min. 2011, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Li, J.; Kim, T.; Nutiu, R.; Ray, D.; Hughes, T.R.; Zhang, Z. Identifying mRNA sequence elements for target recognition by human Argonaute proteins. Genome Res. 2014, 24, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Xia, T.; Santa Lucia, J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic Parameters for an Expanded Nearest-Neighbor Model for Formation of RNA Duplexes with Watson-Crick Base Pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [Green Version]
- Allawi, H.T.; Santa Lucia, J. Thermodynamics and NMR of internal G·T mismatches in DNA. Biochemistry 1997, 36, 10581–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allawi, H.T.; Santa Lucia, J. Thermodynamics of internal C·T mismatches in DNA. Nucleic Acid Res. 1998, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Allawi, H.T.; Santa Lucia, J. Nearest-neighbor thermodynamics of internal A·C mismatches in DNA: Sequence dependence and pH effects. Biochemistry 1998, 37, 9435–9444. [Google Scholar] [CrossRef] [PubMed]
- Allawi, H.T.; Santa Lucia, J. Nearest neighbor thermodynamic parameters for internal G·A mismatches in DNA. Biochemistry 1998, 37, 2170–2179. [Google Scholar] [CrossRef]
- Peyret, N.; Seneviratne, P.A.; Allawi, H.T.; Santa Lucia, J. Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A·A, C·C, G·G, and T·T mismatches. Biochemistry 1999, 38, 3468–3477. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.F.; Holmstrom, E.D.; Nesbitt, D.J. Single-Molecule Kinetics Reveal Cation-Promoted DNA Duplex Formation Through Ordering of Single-Stranded Helices. Biophys. J. 2013, 105, 756–766. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plata, C.A.; Marni, S.; Suweis, S.; Bellini, T.; Paraboschi, E.M. Needles in Haystacks: Understanding the Success of Selective Pairing of Nucleic Acids. Int. J. Mol. Sci. 2022, 23, 3072. https://doi.org/10.3390/ijms23063072
Plata CA, Marni S, Suweis S, Bellini T, Paraboschi EM. Needles in Haystacks: Understanding the Success of Selective Pairing of Nucleic Acids. International Journal of Molecular Sciences. 2022; 23(6):3072. https://doi.org/10.3390/ijms23063072
Chicago/Turabian StylePlata, Carlos A., Stefano Marni, Samir Suweis, Tommaso Bellini, and Elvezia Maria Paraboschi. 2022. "Needles in Haystacks: Understanding the Success of Selective Pairing of Nucleic Acids" International Journal of Molecular Sciences 23, no. 6: 3072. https://doi.org/10.3390/ijms23063072
APA StylePlata, C. A., Marni, S., Suweis, S., Bellini, T., & Paraboschi, E. M. (2022). Needles in Haystacks: Understanding the Success of Selective Pairing of Nucleic Acids. International Journal of Molecular Sciences, 23(6), 3072. https://doi.org/10.3390/ijms23063072