1. Introduction
As a member of the phylum Apicomplexa,
Toxoplasma gondii (
T. gondii) is an obligate intracellular protozoan parasite that can infect almost all warm-blooded animals such as humans and cats. It is estimated that about one third of the world’s human population has been infected with
T. gondii [
1,
2]. Although the infection is usually asymptomatic under normal conditions, it can cause severe complications and even death in immunocompromised individuals. In addition, toxoplasmosis has a severe economic impact on the sheep and goat industries because it induces abortion, stillbirth, and neonatal losses. However, there are no successful
Toxoplasma vaccines that can be applied clinically [
2].
Most of the previous studies focused on rhoptries, micronemes, dense particles, and secretory proteins as virulence or immune effector molecules. However, there has been no significant breakthrough in current research regarding new virulence factors [
3].
T. gondii possesses a highly functional antioxidant system including a group of enzymes such as glutathione peroxidase, superoxide dismutase, catalase, and peroxiredoxins (Prxs), which perform their role independently or synergistically to protect the parasite against adverse conditions
in vivo or
ex vivo [
4]. To date, three
Prx genes have already been identified and characterized functionally and biochemically. Based on the number of active redox cysteine residues and the localization in the cell, the Prxs classified as 1-cys-Prx (TgPrx1) and 2-cys-Prx (TgPrx2) were identified in the cytoplasm of the parasite, while 2-cys-Prx (TgPrx3) was expressed in the mitochondrion, either in tachyzoite or bradyzoite stages [
4]. Peroxiredoxin-linked detoxification of reactive oxygen species plays a critical role in protecting
T. gondii against oxidative stress during its life cycle. This effect makes the peroxiredoxins promising drug targets or vaccine candidates against
T. gondii [
5,
6,
7].
Few reports have investigated the immunogenicity of TgPrxs. A previous study assessed the immunomodulatory role of TgPrx1 derived from the RH strain using an
in vitro model only. The recombinant TgPrx1 enhanced the activity of alternatively activated macrophages (AAM), triggering the production of IL-10 and high expression of arginase-1. Simultaneously, rTgPrx1 induced downregulation of IL-1β secretion from AAM [
8]. Consistently, our previous reports [
9,
10] provided more inclusive data regarding the immunomodulatory role of the recombinant protein of TgPrx1 and TgPrx3. Both rTgPrx1 and rTgPrx3 were investigated as vaccine candidates, and they demonstrated significant protective immunity in immunized mice compared with control non-immunized groups. This protection was accomplished by triggering T-helper 1 and 2 immune responses [
9,
10]. These results prompted us to further assess TgPrx1 and TgPrx3 as vaccine candidates, drug targets, or virulence factors, using knockout techniques as the more recent and reliable approach.
To explore the molecular function of the TgPrx1 and 3 genes, we generated TgPrx1- or TgPrx3-deficient lines of T. gondii type II PruΔku80Δhxgprt (TgPrx1KO and TgPrx3KO), using clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (CRISPR/Cas9). Our results suggest that the phenotype of TgPrx3KO plays a key role in modulating the host’s immunity toward the anti-Toxoplasma activity.
3. Discussion
To date, three
TgPrx genes have been identified in the
T. gondii genome (
TgPrx1,
TgPrx2, and
TgPrx3). The biochemical characterization of
TgPrxs regarding antioxidant enzymes was the most extensively investigated role of such genes [
4,
6,
11]. However, few reports have demonstrated the immunomodulatory effects of TgPrxs including TgPrx1 [
8,
10] and TgPrx3 [
9]. It is noteworthy that most of the above-mentioned reports relied on using recombinant proteins of TgPrx in the biochemical or immunological assessments. The current study provides the first report of the investigation of
TgPrx1 and
TgPrx3 (2-cys peroxiredoxin) using gene deletion or the disruption approach. In the same context, only one previous study has succeeded in deleting
TgPrx2 (1-cys peroxiredoxin) and has also investigated its role as antioxidant enzyme [
12].
Assessment of the protein functions in parasites is primarily based on gene editing or protein expression analyses. Recently, the application of the CRISPR/Cas9 technology has led to great advancements in efficient gene manipulation in apicomplexan parasites including
T. gondii [
12]. The CRISPR/Cas9 system was adapted to produce efficient targeted gene disruption and the site-specific insertions of selectable markers in
T. gondii [
13,
14]. Recently, we succeeded in investigating the role of TgGRA7 and TgGRA14 using CRISPR/Cas9, with a similar approach to that applied in the current study [
15]. This study showed that TgGRA7 and TgGRA14 induced host immunity via NFκB modulation.
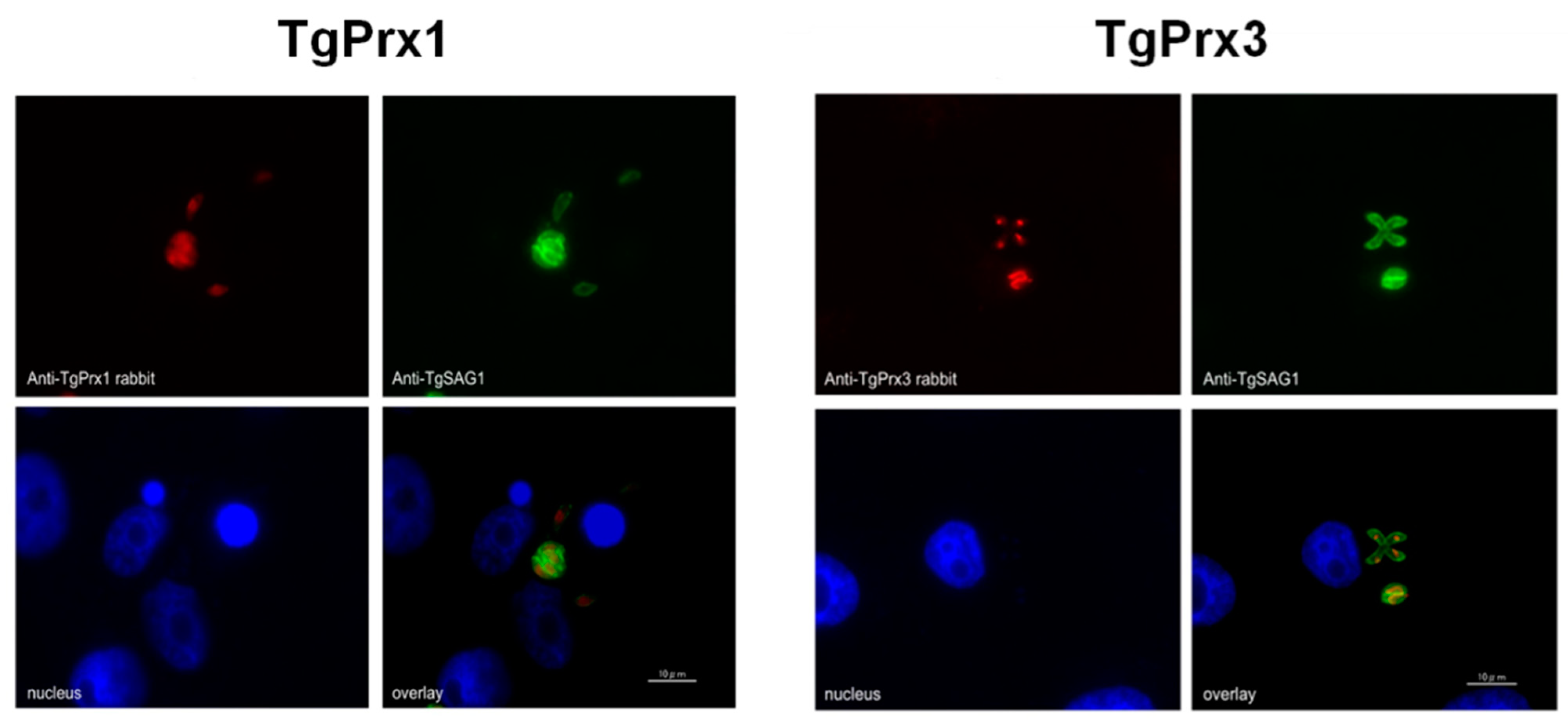
Most previous studies indicated that TgPrx1 was localized in the cytoplasm, while TgPrx3 expression was observed in the mitochondrion [
4,
5,
6]. However, a more recent study referred to a controversial issue by showing evidence of nuclear localization of TgPrx1 in addition to cytosolic expression, using the same antibody [
7]. Data obtained in our study also indicated the cytosolic and nuclear expression of TgPrx1.
In this study, we generated TgPrx1- and TgPrx3-deficient T. gondii PruΔku80Δhxgprt strains to examine the roles of the encoded proteins. The characterization of the specified TgPrx in T. gondii was confirmed by PCR, Western blotting, and IFAT. Two clones from both TgPrx1KO and TgPrx3KO parasites were used to confirm the stability of the obtained data. Correct and precise insertion of the DHFR* cassette was confirmed using different primers to amplify specific sequences from TgPrx and the inserted construct. Western blotting revealed the absence of TgPrx1 and TgPrx3 protein in KO parasites, while it was clearly observed at the expected size in parental strains. In addition, our IFAT results showed the specific localization of TgPrx1 (cyto-nuclear) and TgPrx3 (mitochondrial) in the parental parasites but not the KO parasites. These results indicated the successful gene disruption of both TgPrx1 and TgPrx3.
Based on previous reports, the Prxs genes were expected to be potential drug targets, because of their critical role as antioxidant enzymes that protect the parasites against deleterious oxidative stressors [
5,
16]. Indeed, Conoidin A can bind covalently to the peroxidatic cysteine of TgPrx2, inhibiting its enzymatic activity
in vitro. However, disruption of the
TgPrx2 gene by homologous recombination had no effect on the sensitivity of the parasites to Conoidin A, suggesting that TgPrx2 is not the invasion-relevant target of this compound [
17]. Accordingly, we sought to check the candidacy of TgPrx1 and TgPrx3, as drug targets and for the discovery of specific inhibitors. However, the disruption of both
TgPrx1 and
TgPrx3 did not affect the abilities of the parasite with regard to infection, replication, or egress
in vitro using Vero cells. In this experiment, two clones from each KO parasite (
TgPrx1KO and
TgPrx3KO) were used to check the data stability. Furthermore, the mice infection experiments abolished our strategy to check TgPrx1 and TgPrx3 as drug targets due to the increased or similar virulence against the parental parasite in
TgPrx3KO and
TgPrx1KO, respectively. This result was confirmed using various mouse models including different mouse strains and sexes.
However, many previous reports demonstrated the similar phenomenon of increased parasite virulence after deletion of certain genes. Deletion of TgGRA7, TgGRA14, and TgGRA15 were demonstrated to increase the virulence of the parasites, in the case of either type I RHΔku80Δhxgprt or type II PruΔku80Δhxgprt strains [
15]. Similarly, mice infected with TgGRA15KO parasites showed a higher parasite burden and higher growth rate
in vitro than those infected with the parental parasite [
18]. Furthermore, a deficiency of aspartate aminotransferases in both RH and PLK did not reduce the virulence in mice, although the growth ability of the parasites was affected
in vitro [
19]. This phenomenon demonstrates the close co-evolution of
T. gondii and host.
T. gondii evolved in ways that preserved the host’s wellbeing up to a point, avoiding extreme damage to the host and ensuring parasite survival and continuation of the life cycle.
Toxoplasma gondii has a fascinating ability to manipulate the effector immune cells and molecules to maintain its survival and to keep the host alive. Both cellular and humoral immunity is involved in combating the parasite. Macrophages are the first line of host defense against many infectious agents including
T. gondii. They are responsible for the production of a vast group of cytokines that are essential for regulating the immune response against invading
T. gondii. In the early stage of infection, classically activated macrophages can produce a group of proinflammatory cytokines such as interleukin 6 (IL-6), IL-12, and tumor necrosis factor alpha (TNF-α). In addition, IL-12 can promote the function of natural killer cells and T-cells to produce interferon gamma (IFN-γ). These cytokines can act synergistically to mediate the killing of the parasite by macrophages [
20,
21,
22].
The increased virulence of the TgPrx3KO parasite in mice may suggest the immunoprotective role of such antigens during infection. However, the deletion of the TgPrx1 gene did not affect the parasite phenotype either in vitro or in vivo, as found for TgPrx3 in the current study. In fact, both TgPrx1 and TgPrx3 are 2-cys peroxiredoxins, but they have different localizations of expression, so various effects might be expected.
To identify the mechanism of the protective role of TgPrx3 during infection, the IFN-γ level was measured as a marker of sickness in serum and ascites of mice at 8 dpi, which was the day on which severe infection was observed. We found that mice infected with both
TgPrx3KO and
TgPrx1KO parasites showed higher IFN-γ secretion than those infected with the parental PruΔku80Δhxgprt strain. However, in another experiment, higher levels of proinflammatory cytokines IL-6 and IL-12p40 were observed mainly in
TgPrx3KO parasites against both
TgPrx1KO- and PruΔku80Δhxgprt-infected macrophages. These results may explain the high severity of infection in the case of
TgPrx3KO-infected mice. Interestingly, this result was different from those we obtained previously, as both recombinant TgPrx1 [
10] and rTgPrx3 [
9] triggered the IL-12 production from macrophages. However, this different effect might be correlated with the different approaches of KO and recombinant protein assays. In the case of parasite infection, there is an intricate effect of numerous protein–protein interactions, even for the parasite itself, rather than using one antigen as in the recombinant protein assessment approach. However, our previous studies reported that the use of rTgPrx1 and rTgPrx3 as vaccine antigens promoted cellular and humoral immune responses and improved mouse survival [
9,
10]. These results corroborate the findings of the current study concerning the efficient role of TgPrx1 and TgPrx3 in manipulating host immunity towards protecting the host against the damaging effect of the parasite. Another aspect of the induced high virulence of parasites lacking
TgPrx3 may be related to its potential role as an antioxidant enzyme. This antioxidant effect was not investigated in the current study, but it is the most frequently investigated aspect in all identified Prxs of
T. gondii including
TgPrx3 [
4,
6,
16]. Oxidative stress caused by accumulation of reactive oxygen species (ROS) such as hydrogen peroxide and nitric oxide has a highly harmful effect on the host cells either
in vivo or
in vitro. The infection with
T. gondii resulted in the production of high levels of a variety of ROS triggered by the activation of macrophages or other immune cells [
23,
24].
In this study, we confirmed the successful knockout of TgPrx1 and TgPrx3 using different experimental approaches (PCR, Western blotting, and IFAT). In addition, two clones from each KO parasite were used in the experiments to corroborate the obtained data and the effect of gene deletion. However, our efforts to generate complementary parasites for TgPrx1KO or TgPrx3KO were not successful, suggesting further attempts to investigate the role of TgPrx1 and TgPrx3 are necessary, using a similar approach. These findings can be exploited in the further assessment of TgPrx3 as a vaccine antigen against T. gondii using various animal models and different vaccination strategies.
4. Materials and Methods
4.1. Ethics Statement
The current study was conducted strictly according to the recommendations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The protocol was approved by the Committee on the Ethics of Animal Experiments at Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan (permit numbers 20-22, 19-50, 18-38, 29-39). All painful experiments were conducted under isoflurane anesthesia, and every effort was made to minimize animal suffering.
4.2. Animals
Female and male ICR, male C57BL/6 and SCID mice, and female BALB/c mice, at 6 to 8 weeks of age, were obtained from Clea Japan (Tokyo, Japan). These mice were housed under specific-pathogen-free conditions in cages in the animal facility of the National Research Center for Protozoan Diseases at Obihiro University of Agriculture and Veterinary Medicine, Obihiro, Japan. The animals used in this study were treated and used according to the Guiding Principles for the Care and Use of Research Animals published by the Obihiro University of Agriculture and Veterinary Medicine.
4.3. Parasite and Cell Cultures
All the parasite strains used in this study (T. gondii type II PruΔku80Δhxgprt, TgPrx1KO, and TgPrx3KO) were maintained in African green monkey kidney epithelial cells (Vero cells) cultured in Eagle’s minimum essential medium (Sigma, St. Louis, MO, USA) containing 8% heat-inactivated fetal bovine serum (FBS) in a 37 °C and 5% CO2 incubator. T. gondii type II PruΔku80Δhxgprt was kindly gifted by Professor David Bzik (Dartmouth Medical School). Prior to each experiment, the parasites were purified from host cell debris and washed with cold phosphate-buffered saline (PBS). Finally, the pellet was resuspended carefully in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma, St. Louis, MO, USA) and passed through a 27-gauge needle and a 5.0 µm pore size filter (Millipore, Bedford, MA, USA).
4.4. Plasmid Construction and Generation of Knockout Parasites
The
TgPrx1 and
TgPrx3 gene knockout strains were constructed as described in the literature, using CRISPR-Cas9 [
13,
15,
25]. SgRNA from
TgPrx1 and
TgPrx3 was briefly transmitted into pSAG1:CAS9-U6:sgUPRT by PCR using a Q5 Mutagenesis Kit, and positive plasmid of pSAG1:CAS9-U6:sg
TgPrx1 or
TgPrx3 was extracted using Endo-Free Plasmid DNA Mini Kit protocols (Takara, Kusatsu, Shiga, Japan). The dihydrofolate reductase (DHFR) resistance cassettes were amplified from the plasmid pUPRT-DHFR-D (Addgene, Watertown, MA, USA) by PCR reaction and purified by agarose gel electrophoresis. About 50 μg of positive plasmids and 5 μg of purified DHFR amplicons were cotransfected into freshly harvested PruΔku80Δhxgprt tachyzoites by electroporation, as described previously [
25]. Stably resistant clones were selected by growth in pyrimethamine (1 µM) for 14 days and were subsequently screened via PCR to ensure the correct integration of the DHFR cassette into each target gene locus (see
Figures S1 and S2 and Table S1 in the Supplementary Material. The PCR-positive clones were further analyzed via Western blotting and the indirect fluorescent antibody test (IFAT) to confirm the loss of the target gene. Two clones from each parasite were identified and confirmed, named
TgPrx1KO1,
TgPrx1KO2,
TgPrx3KO2, and
TgPrx3KO5.
4.5. Expression and Purification of Recombinant Proteins
The
TgPrx1 and
TgPrx3 genes were amplified from cDNA of the
T.
gondii PLK strain, as described in our previous studies [
9,
10]. In brief, PCR products digested with Bam
HI and
XhoI were inserted into the pGEX-4T3 plasmid vector (
Table S1). Recombinant TgPrx1 and TgPrx3 were expressed as glutathione
S-transferase (GST) fusion protein in
Escherichia coli BL21(DE3) (New England Biolabs Inc., Ipswich, MA, USA).
4.6. Preparation of Lysate Antigen
Lysate antigen from purified tachyzoites of
T. gondii (PruΔku80Δhxgprt),
TgPrx1KO, or
TgPrx3KO was prepared as previously reported [
10]. The obtained extract was measured using a BCA protein assay kit to detect the concentrations (Thermo Fisher Scientific, Waltham, MA, USA).
4.7. Western Blot Analysis
The protein lysates from purified
T.
gondii tachyzoites (2 or 5 μg/10 μL for
TgPrx1KO and
TgPrx3KO, respectively) were mixed with 10 μL of 2×SDS reducing gel-loading buffer (62.5 mM Tris-HCl pH 6.8, 2% (
w/
v) SDS, 140 mM 2-mercaptoethanol, 10% (
w/
v) glycerol and 0.02% (
w/
v) bromophenol blue). Other procedures were applied as described in our previous report [
10]. Briefly, samples were heated at 95°C for 5 min and separated on a 15% polyacrylamide gel. The membranes were incubated with mouse anti-TgSAG1 (monoclonal antibody F77D; Waltham, MA, USA), or anti-TgPrx1 [
10] or anti-TgPrx3 [
9] mouse serum (1:200) for 1 h at room temperature. After washing three times, the membranes were incubated with anti-mouse horseradish peroxidase-conjugated immunoglobulin G (1:2000; Amersham Pharmacia Biotech, Piscataway, NJ, USA) diluted in PBS–skim milk (3%), for 1 h at 37 °C. The protein bands were visualized using ECL™ Western blotting detection reagents (GE Healthcare UK Ltd., Buckinghamshire, UK) using a VersaDoc™ imaging system (Nippon Bio-Rad Laboratories, Tokyo, Japan) according to the manufacturer’s recommendations.
4.8. Indirect Fluorescent Antibody Test (IFAT)
A Vero cells suspension of 5 × 10
4 cells in 1 mL of MEM supplemented with 8% FBS was seeded in each well in a 12-well plate with a coverslip and incubated at 37 °C in a 5% CO
2 atmosphere for 24 h. The cells were infected with purified tachyzoites of different parasite lines at 2 × 10
5/1 mL of MEM and incubated again at 37 °C in a 5% CO
2 atmosphere for 2 h, followed by washing with PBS to remove dead parasites. Plates were incubated again under the same conditions for 48 h for characterization experiments, 24 h for infection rate, 48 h for proliferation rate, and 72 h for egress rate assessment [
15,
25]. Old medium was aspirated, and coverslips were washed with PBS 3 times, followed by fixation with 3% paraformaldehyde in PBS (
v/v) for 30 min at RT. After washing 2 more times with PBS, permeation of cells was achieved by adding 0.3% Triton X-100 in PBS and keeping for 5 min at RT. The coverslips were washed 3 more times followed by blocking with 3% bovine serum albumin (BSA) in PBS (BSA–PBS) for 30 min at RT. As for the first antibodies, cells were stained by fluorescent-labeled antibodies; anti-mouse TgSAG1 monoclonal antibodies (Invitrogen) (1:500) or anti-rabbit TgPrx1 [
10] or TgPrx3 [
9] polyclonal antibodies (1:200). For secondary anti-mouse or anti-rabbit antibodies, Alexa Fluor (1:500 dilution, Sigma, St. Louis, MO, USA) and Hoechst (1:1000, Thermo Fisher Scientific Inc., Waltham, MA, USA) were used as green or red staining, depending on the type of antibodies used, diluted 1:500 in 3% BSA–PBS for 1 h at RT. The coverslips were placed on a glass slide containing a fresh drop of Mowiol (Calbiochem, San Diego, CA, USA), and the slides kept in the dark for at least 2 h. Slides were examined using an all-in-one fluorescence microscope (BZ-9000, Keyence, Tokyo, Japan).
4.9. Infection, Growth, and Egress Rates of T. gondii Lines
Vero cells were plated at 1 mL/well in a 12-well plate with a coverslip by suspension, at 5 × 10
4 in MEM 8% FBS, then incubated for 20 h at 37 °C. Parasites were plated in at 1 × 10
5 in MEM using 1 mL/well (multiplicity of infection 1:4 host to parasite cells). The parasites that failed to invade cells were washed away, and the cultures were incubated again as previously mentioned. Counting, calculation, and analyses were performed as described previously [
15]. After 24 h, 48 h, or 72 h, the cells were fixed by paraformaldehyde (3%) in PBS and prepared for IFAT for detection of infection, proliferation, or egress rates, respectively. The infection rates were calculated using IFAT as follows: (number of TgSAG1-positive Vero cells/100 randomly selected Vero cells) × 100. To measure the
T. gondii proliferation rate in the Vero cells, the sizes of the parasitophorous vacuoles (PVs) were determined by counting the number of parasites per PV (in a total of 100 randomly selected vacuoles) and expressed as a percentage (%) of the total PV based on the TgSAG1 fluorescent signals. In the case of egress rate, i.e., parasite egress in the Vero cells, the percentage of egressed vacuoles was calculated by scoring at least 100 vacuoles as intracellular or egressed, relying on TgSAG1 fluorescent signals. All precautions were applied to avoid any errors and duplicate counting. Additionally, all assessment procedures for infection, replication, or egress experiments were conducted blindly by a neutral person by hiding the slide ID.
4.10. In Vivo Assessment of PruΔku80Δhxgprt and TgPrxKO Strains in Mice
To compare the virulence of T. gondii in mice, female and male ICR, male C57BL/6, and male SCID mice were intraperitoneally injected with different parasite lines of T. gondii (5 × 104 tachyzoites/mouse). The mouse survival was assessed in all trials of different mice (30 days post infection (dpi) in female and 70 dpi in male mice). In addition, body weight and clinical score were monitored daily for up to 30 dpi, and the sera and brains of female ICR mice were collected at the sacrifice time of 30 dpi. To analyze cytokine production in vivo, sera and ascites were collected from the female mice infected with PruΔku80Δhxgprt, TgPrx1KO1, or TgPrx3KO2 strains or inoculated with RPMI medium only (n = 4 per group) at 8 dpi.
4.11. Monolayer Cultures of Peritoneal Macrophages
Peritoneal macrophages from female ICR mice were collected 4 days after the intraperitoneal injection of 1 mL of 4.05% Brewer’s modified thioglycollate medium (BBL) (Becton, Dickinson and Company, Sparks, MD, USA), as described previously [
10]. A macrophage suspension (4 × 10
5) prepared in Dulbecco’s modified Eagle’s medium (DMEM; Sigma, St. Louis, MO, USA) supplemented with 10% heat-inactivated FBS was added to 96-well tissue culture microplates. The suspension was incubated at 37 °C for 3 h, washed thoroughly to remove nonadherent cells, and incubated again at 37 °C for 24 h. Parasite suspensions in 200 μL of DMEM (2.5 × 10
4/well, 5 × 10
4/well, 1 × 10
5/well) were added to each well. At 2 h post infection, the extracellular parasites were washed away and DMEM supplemented with 10% FBS was added. At 20 h post infection, the culture supernatants (150 μL from each well) were collected for the measurement of cytokines.
4.12. Sandwich ELISA for Measuring Cytokine Levels
The level of IFN-γ in the sera and ascites fluid and of IL-6 and IL-12p40 in the macrophage culture supernatant were determined via commercial sandwich ELISAs (Pierce Biotechnology Inc., Rockford, IL, USA), according to the manufacturer’s instructions. The standard cytokine curves constructed from the samples run on the same plate were used for the calculation of cytokine concentrations.
4.13. Statistical Analysis
All statistical analyses were performed using GraphPad Prism version 5 (GraphPad Software Inc., La Jolla, CA, USA). Statistical analyses were performed using a one-way analysis of variance (ANOVA) followed by the Tukey–Kramer test for group comparisons in analyzing rates of infection, proliferation, and egress of parasites, and for cytokine production, with the data for each presented as mean values ± standard deviations. The significance of differences in mouse survival was analyzed using a log rank test, and p-values < 0.05 were considered statistically significant.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}