
Exosomal Carboxypeptidase E (CPE) and CPE-shRNA-Loaded Exosomes Regulate Metastatic Phenotype of Tumor Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
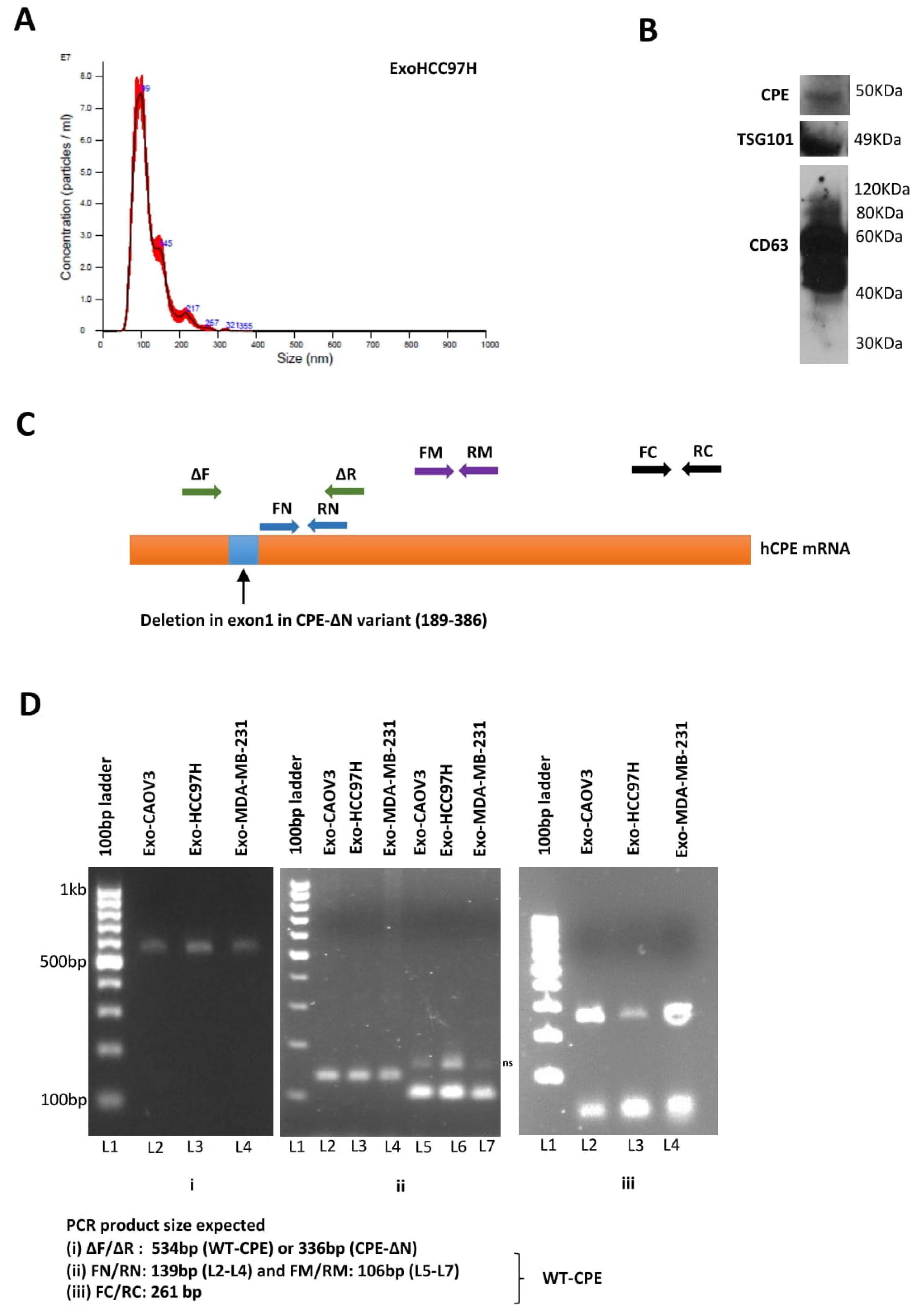
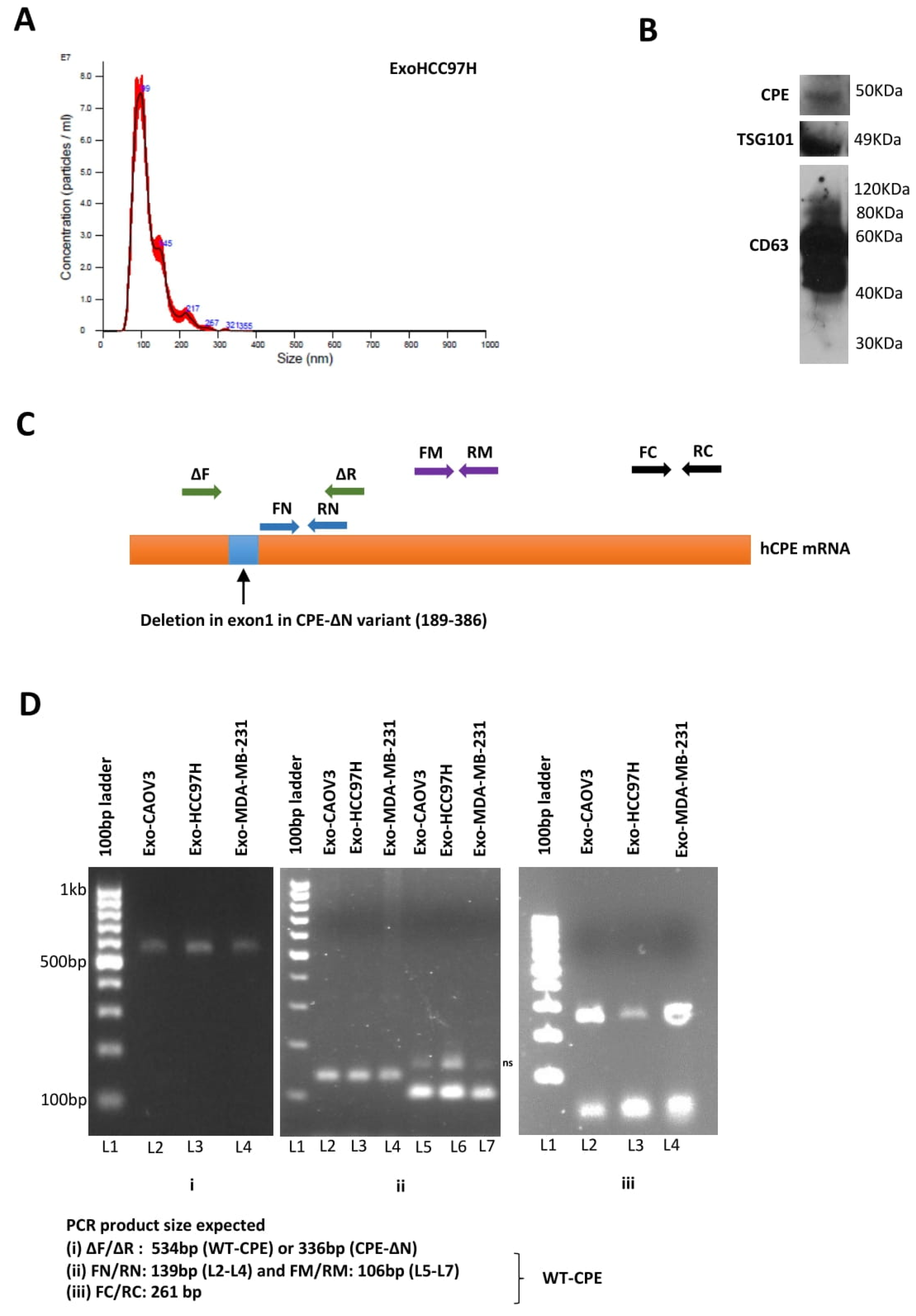
2.1. Presence of CPE in Exosomes Derived from Cancer Cells
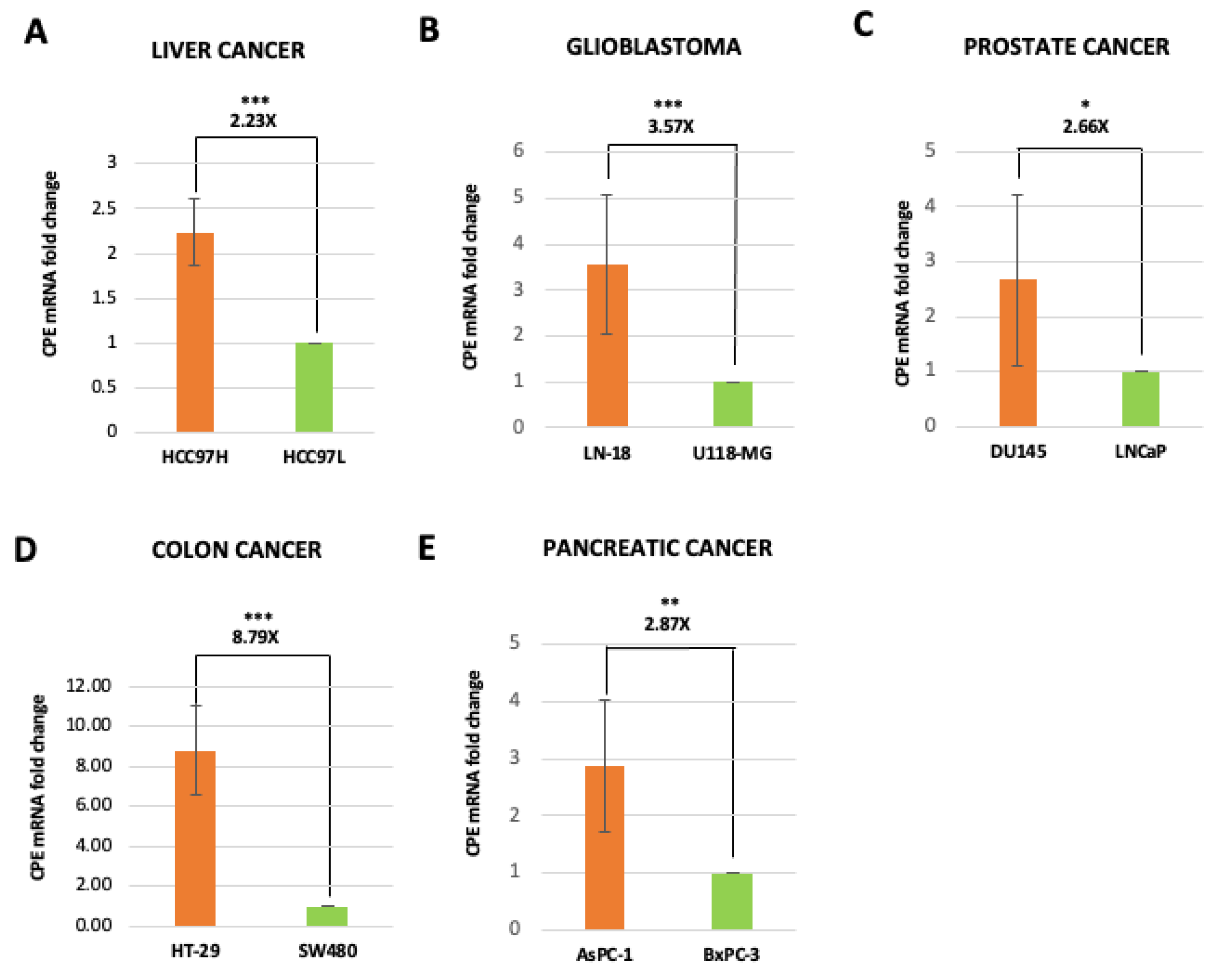
2.2. Exosomes Isolated from Highly Malignant Cancer Cells Show Elevated CPE mRNA Levels
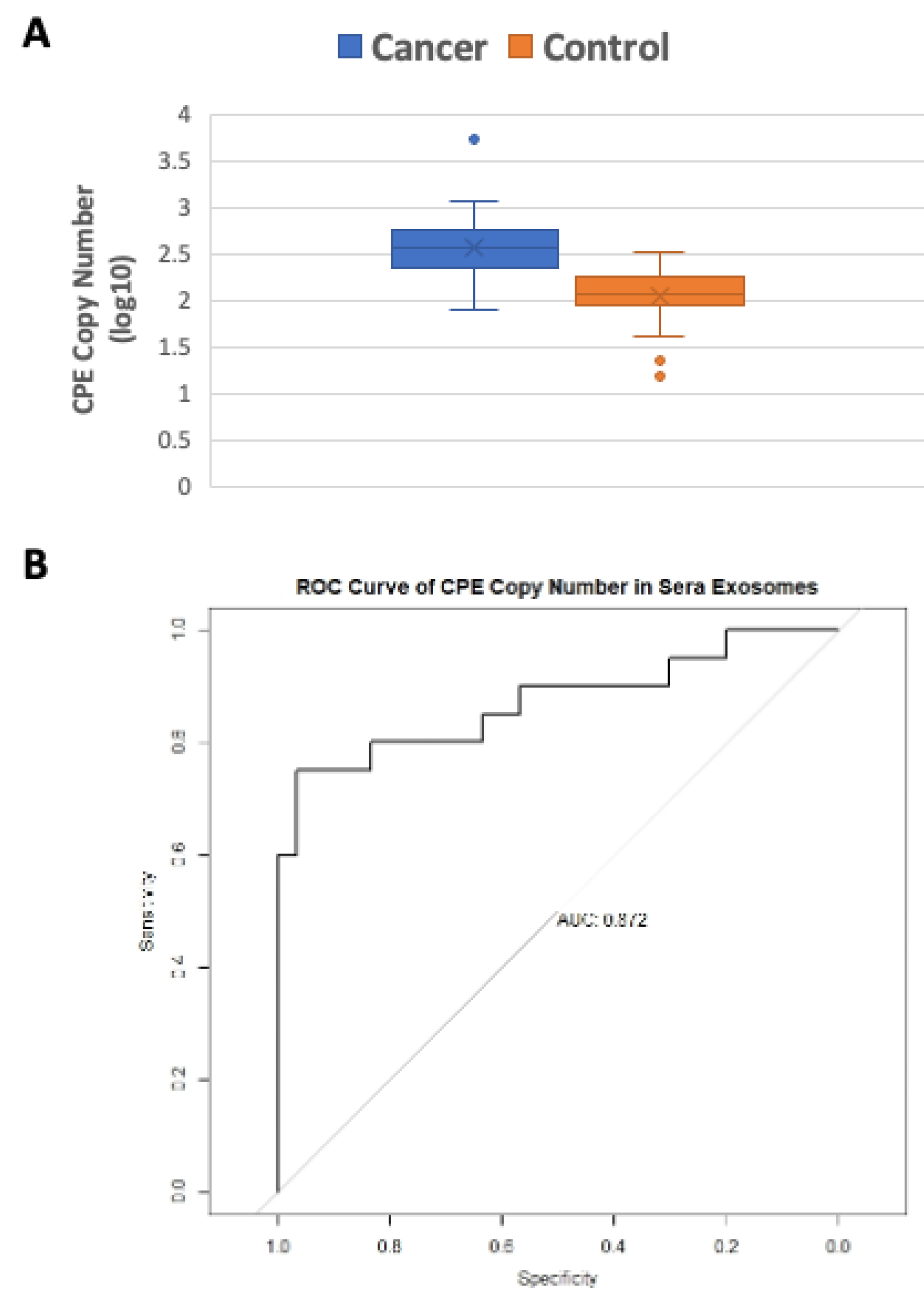
2.3. Serum Exosomes from Cancer Patients Have Higher CPE Transcript Copy Numbers than Healthy Controls
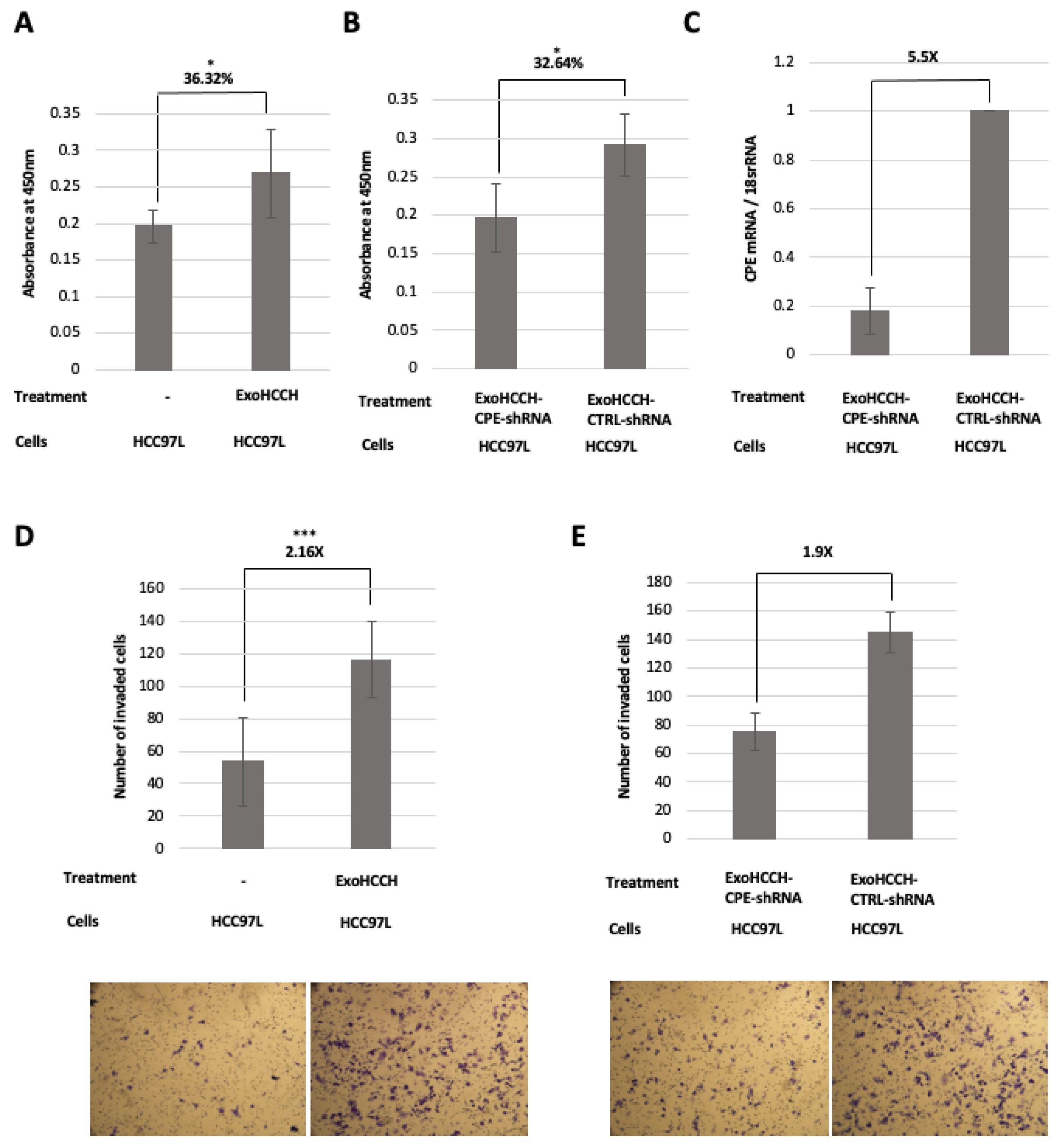
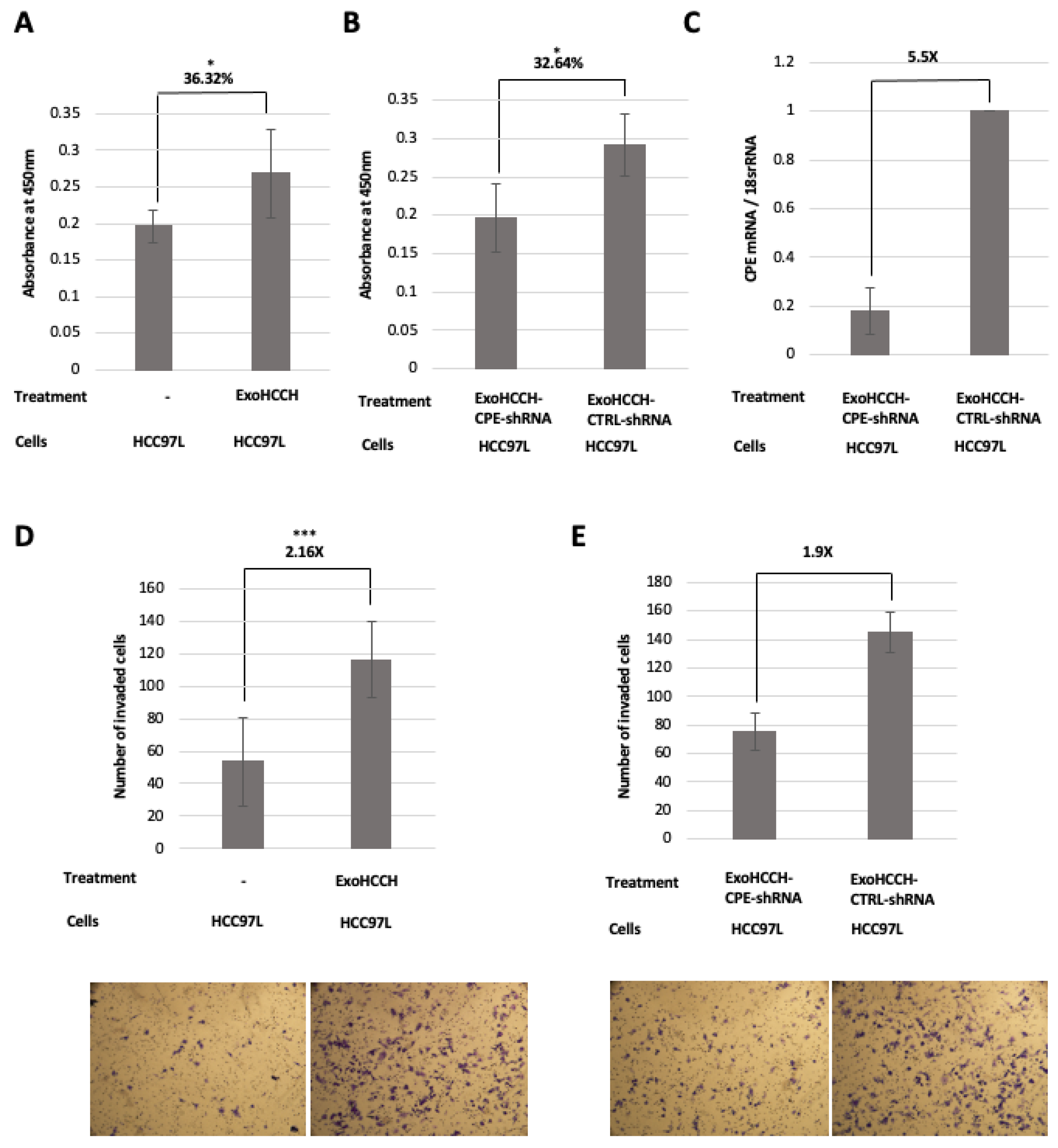
2.4. HCC97H Exosomes Enhance Proliferation and Invasion of HCC97L Cells in a CPE-Dependent Manner
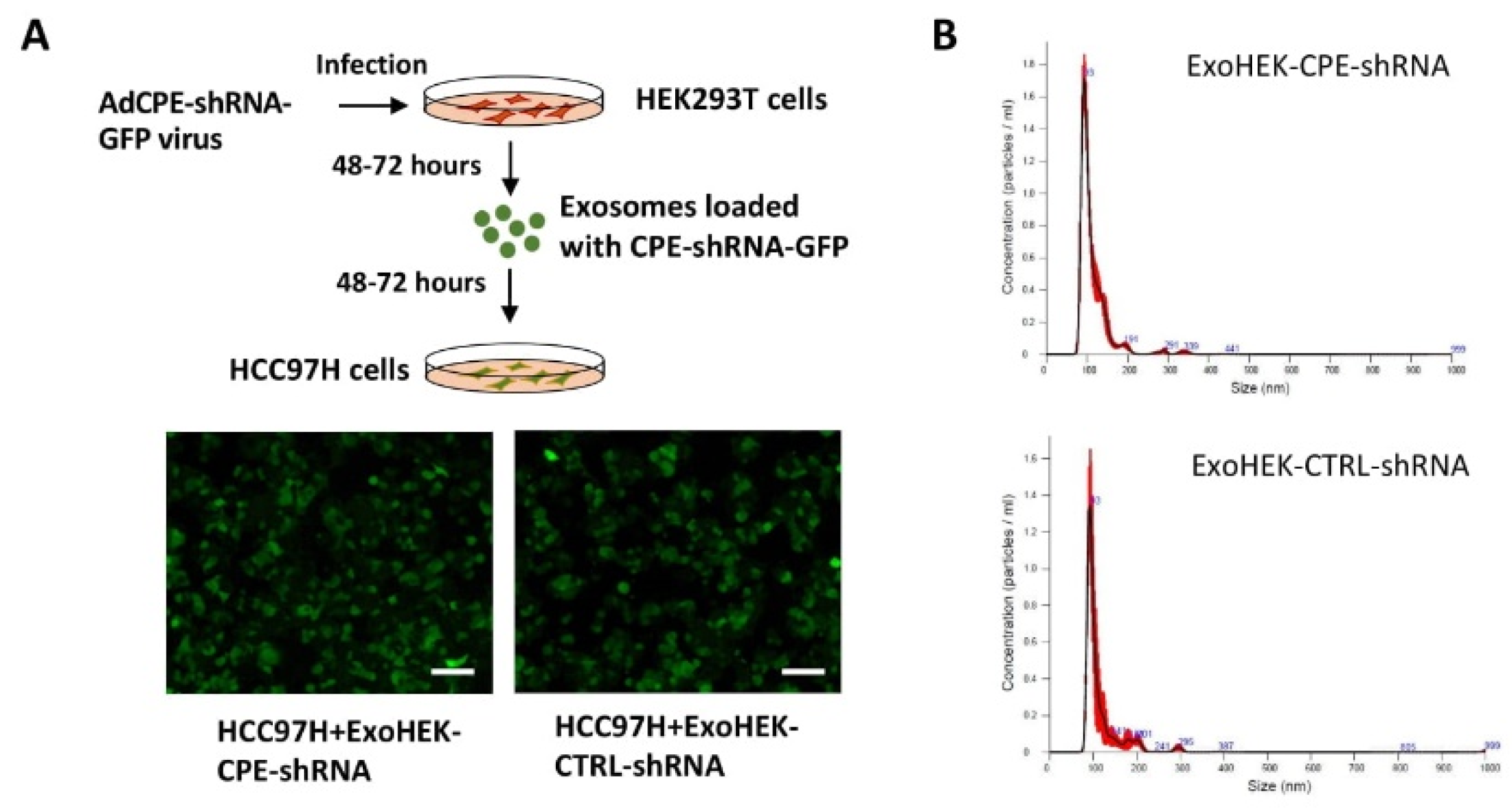
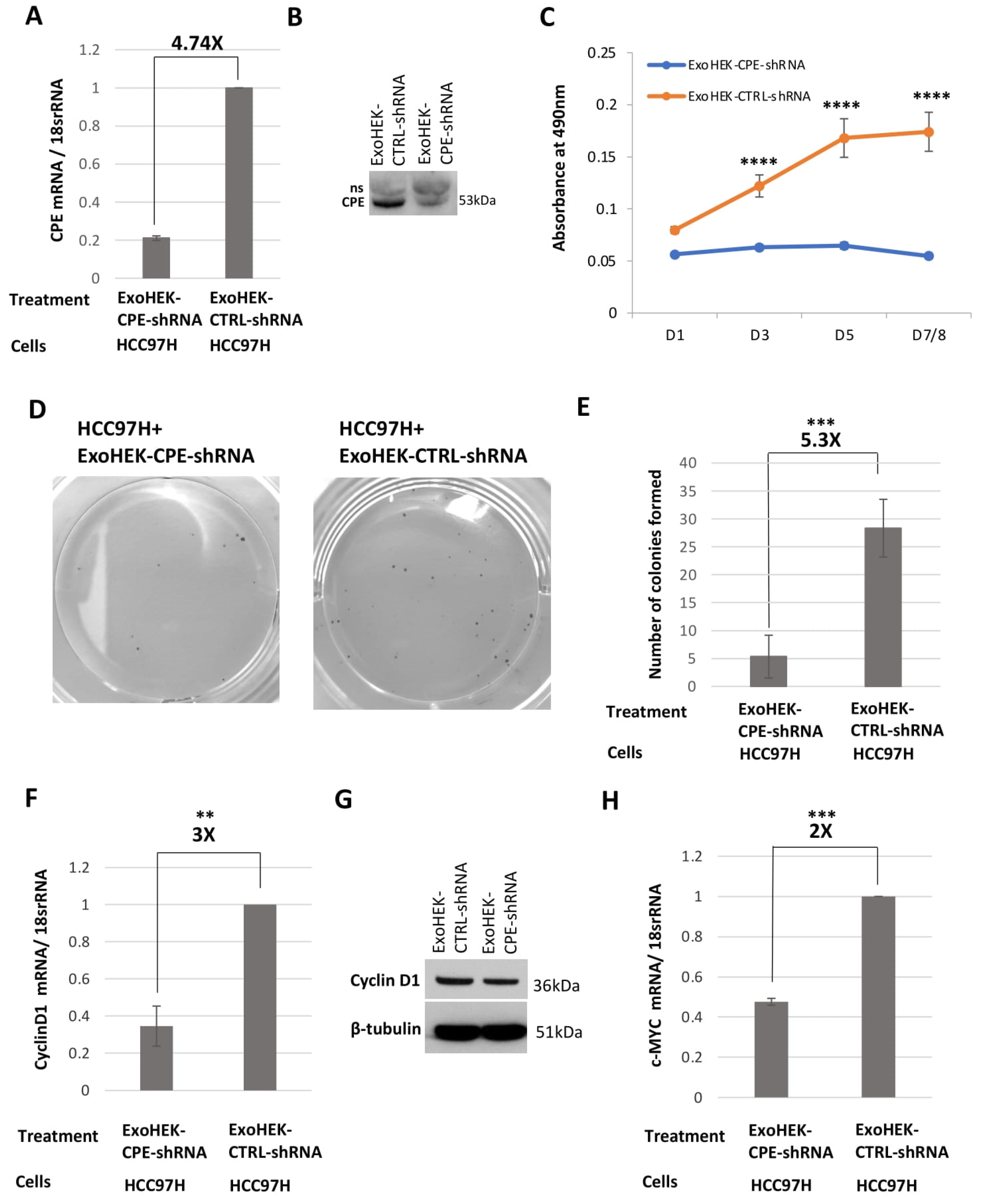
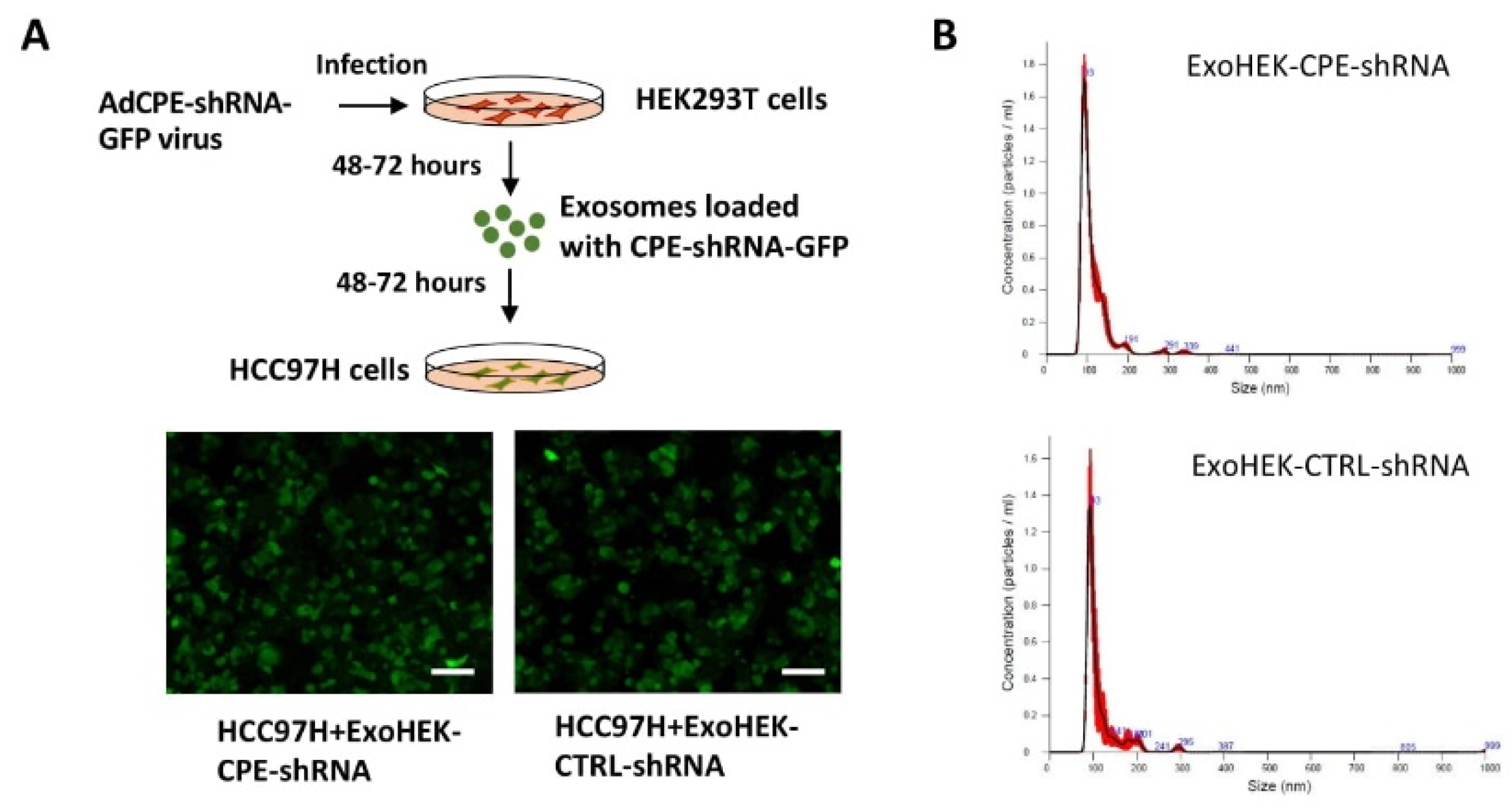
2.5. Exosomes Loaded with CPE-shRNA Inhibit Proliferation of Malignant HCC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Patient Serum Samples
4.3. Isolation of Exosomes
4.4. NanoSight Analysis
4.5. RT-PCR
4.6. Quantitative Real-Time PCR
4.7. Western Blot
4.8. In Vitro Exosome Transfer Experiments
4.9. Cell Proliferation Assay
4.10. Matrigel Invasion Assay
4.11. Colony Formation Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPE | Carboxypeptidase E |
| HCC | Hepatocellular carcinoma |
| HEK | Human embryonic kidney |
| TSG101 | Tumor susceptibility gene 101 |
| AUC | Area under the curve |
| ROC | Receiver operating characteristics |
| GFP | Green fluorescent protein |
| NTA | Nanoparticle tracking analysis |
| EMT | Epithelial-Mesenchymal Transition |
References
- Tamura, T.; Yoshioka, Y.; Sakamoto, S.; Ichikawa, T.; Ochiya, T. Extracellular vesicles as a promising biomarker resource in liquid biopsy for cancer. Extracell. Vesicles Circ. Nucleic Acids 2021, 2, 148–174. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luga, V.; Wrana, J.L. Tumor-stroma interaction: Revealing fibroblast-secreted exosomes as potent regulators of Wnt-planar cell polarity signaling in cancer metastasis. Cancer Res. 2013, 73, 6843–6847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Guo, P.; He, Y.; Chen, Z.; Chen, L.; Luo, Y.; Qi, L.; Liu, Y.; Wu, Q.; Cui, Y.; et al. HCC-derived exosomes elicit HCC progression and recurrence by epithelial-mesenchymal transition through MAPK/ERK signalling pathway. Cell Death Dis. 2018, 9, 513. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Feng, X.; Liu, H.; Tong, R.; Wu, J.; Li, C.; Yu, H.; Chen, Y.; Cheng, Q.; Chen, J.; et al. High-metastatic cancer cells derived exosomal miR92a-3p promotes epithelial-mesenchymal transition and metastasis of low-metastatic cancer cells by regulating PTEN/Akt pathway in hepatocellular carcinoma. Oncogene 2020, 39, 6529–6543. [Google Scholar] [CrossRef]
- Fu, Q.; Zhang, Q.; Lou, Y.; Yang, J.; Nie, G.; Chen, Q.; Chen, Y.; Zhang, J.; Wang, J.; Wei, T.; et al. Primary tumor-derived exosomes facilitate metastasis by regulating adhesion of circulating tumor cells via SMAD3 in liver cancer. Oncogene 2018, 37, 6105–6118. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Liu, W.; Zou, Y.; Wang, G.; Deng, Y.; Luo, J.; Zhang, Y.; Li, H.; Zhang, Q.; Yang, Y.; et al. Three isoforms of exosomal circPTGR1 promote hepatocellular carcinoma metastasis via the miR449a-MET pathway. EBioMedicine 2019, 40, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Kia, V.; Paryan, M.; Mortazavi, Y.; Biglari, A.; Mohammadi-Yeganeh, S. Evaluation of exosomal miR-9 and miR-155 targeting PTEN and DUSP14 in highly metastatic breast cancer and their effect on low metastatic cells. J. Cell. Biochem. 2019, 120, 5666–5676. [Google Scholar] [CrossRef]
- Shen, X.; Wang, C.; Zhu, H.; Wang, Y.; Wang, X.; Cheng, X.; Ge, W.; Lu, W. Exosome-mediated transfer of CD44 from high-metastatic ovarian cancer cells promotes migration and invasion of low-metastatic ovarian cancer cells. J. Ovarian Res. 2021, 14, 38. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Lee, Y.; Lakhal-Littleton, S.; Li, J.; Seow, Y.; Gardiner, C.; Alvarez-Erviti, L.; Sargent, I.L.; Wood, M.J. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat. Protoc. 2012, 7, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Fais, S. Exosomes: A Source for New and Old Biomarkers in Cancer. Cancers 2020, 12, 2566. [Google Scholar] [CrossRef] [PubMed]
- Frampton, A.E.; Prado, M.M.; Lopez-Jimenez, E.; Fajardo-Puerta, A.B.; Jawad, Z.A.R.; Lawton, P.; Giovannetti, E.; Habib, N.A.; Castellano, L.; Stebbing, J.; et al. Glypican-1 is enriched in circulating-exosomes in pancreatic cancer and correlates with tumor burden. Oncotarget 2018, 9, 19006–19013. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Y.; Qiu, F.; Qiu, Z. Proteomic identification of exosomal LRG1: A potential urinary biomarker for detecting NSCLC. Electrophoresis 2011, 32, 1976–1983. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal miRNAs as Biomarkers for Prostate Cancer. Front. Genet. 2013, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Wani, S.; Kaul, D.; Mavuduru, R.S.; Kakkar, N.; Bhatia, A. Urinary-exosomal miR-2909: A novel pathognomonic trait of prostate cancer severity. J. Biotechnol. 2017, 259, 135–139. [Google Scholar] [CrossRef]
- Li, Z.; Ma, Y.Y.; Wang, J.; Zeng, X.F.; Li, R.; Kang, W.; Hao, X.K. Exosomal microRNA-141 is upregulated in the serum of prostate cancer patients. OncoTargets Ther. 2016, 9, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Fricker, L.D.; Snyder, S.H. Purification and characterization of enkephalin convertase, an enkephalin-synthesizing carboxypeptidase. J. Biol. Chem. 1983, 258, 10950–10955. [Google Scholar] [CrossRef]
- Hook, V.Y.; Loh, Y.P. Carboxypeptidase B-like converting enzyme activity in secretory granules of rat pituitary. Proc. Natl. Acad. Sci. USA 1984, 81, 2776–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Cawley, N.X.; Loh, Y.P. Carboxypeptidase E/NFalpha1: A new neurotrophic factor against oxidative stress-induced apoptotic cell death mediated by ERK and PI3-K/AKT pathways. PLoS ONE 2013, 8, e71578. [Google Scholar] [CrossRef] [Green Version]
- Cool, D.R.; Normant, E.; Shen, F.; Chen, H.C.; Pannell, L.; Zhang, Y.; Loh, Y.P. Carboxypeptidase E is a regulated secretory pathway sorting receptor: Genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell 1997, 88, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Dhanvantari, S.; Loh, Y.P. Lipid raft association of carboxypeptidase E is necessary for its function as a regulated secretory pathway sorting receptor. J. Biol. Chem. 2000, 275, 29887–29893. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.G.; Musat, M.; Czirjak, S.; Hanzely, Z.; Lillington, D.M.; Korbonits, M.; Grossman, A.B. Differential gene expression in pituitary adenomas by oligonucleotide array analysis. Eur. J. Endocrinol. 2005, 153, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Armento, A.; Ilina, E.I.; Kaoma, T.; Muller, A.; Vallar, L.; Niclou, S.P.; Kruger, M.A.; Mittelbronn, M.; Naumann, U. Carboxypeptidase E transmits its anti-migratory function in glioma cells via transcriptional regulation of cell architecture and motility regulating factors. Int. J. Oncol. 2017, 51, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.F.; Wu, H.D.; Chen, Y.T.; Murthy, S.R.; Chiu, Y.T.; Chang, Y.; Chang, I.C.; Yang, X.; Loh, Y.P. Carboxypeptidase E is a prediction marker for tumor recurrence in early-stage hepatocellular carcinoma. Tumour Biol. 2016, 37, 9745–9753. [Google Scholar] [CrossRef]
- Liu, A.; Shao, C.; Jin, G.; Liu, R.; Hao, J.; Shao, Z.; Liu, Q.; Hu, X. Downregulation of CPE regulates cell proliferation and chemosensitivity in pancreatic cancer. Tumour Biol. 2014, 35, 12459–12465. [Google Scholar] [CrossRef]
- Shen, H.W.; Tan, J.F.; Shang, J.H.; Hou, M.Z.; Liu, J.; He, L.; Yao, S.Z.; He, S.Y. CPE overexpression is correlated with pelvic lymph node metastasis and poor prognosis in patients with early-stage cervical cancer. Arch. Gynecol. Obstet. 2016, 294, 333–342. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Faddaoui, A.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Guillemette, C.; Gobeil, S.; Macdonald, E.; et al. BCAT1 expression associates with ovarian cancer progression: Possible implications in altered disease metabolism. Oncotarget 2015, 6, 31522–31543. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Li, X.; Li, L.; Wang, L.; Du, Z.; Yang, Y.; Zhao, J.; Li, Y. Silencing of carboxypeptidase E inhibits cell proliferation, tumorigenicity, and metastasis of osteosarcoma cells. OncoTargets Ther. 2016, 9, 2795–2803. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Li, L.L.; Wu, G.G.; Xie, Y.C.; Zhang, G.X.; Chen, W.; Yang, H.F.; Liu, Q.L.; Li, W.H.; He, W.G.; et al. Upregulation of CPE promotes cell proliferation and tumorigenicity in colorectal cancer. BMC Cancer 2013, 13, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hareendran, S.; Yang, X.; Lou, H.; Xiao, L.; Loh, Y.P. Carboxypeptidase E-N Promotes Proliferation and Invasion of Pancreatic Cancer Cells via Upregulation of CXCR2 Gene Expression. Int. J. Mol. Sci. 2019, 20, 5725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lou, H.; Chen, Y.T.; Huang, S.F.; Loh, Y.P. A novel 40kDa N-terminal truncated carboxypeptidase E splice variant: Cloning, cDNA sequence analysis and role in regulation of metastatic genes in human cancers. Genes Cancer 2019, 10, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tang, Z.Y.; Ye, S.L.; Liu, Y.K.; Chen, J.; Xue, Q.; Chen, J.; Gao, D.M.; Bao, W.H. Establishment of cell clones with different metastatic potential from the metastatic hepatocellular carcinoma cell line MHCC97. World J. Gastroenterol. 2001, 7, 630–636. [Google Scholar] [CrossRef]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabro, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.; Skog, J.; Nordstrand, A.; Baranov, V.; Mincheva-Nilsson, L.; Breakefield, X.O.; Widmark, A. Prostate cancer-derived urine exosomes: A novel approach to biomarkers for prostate cancer. Br. J. Cancer 2009, 100, 1603–1607. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kamohara, H.; Kinoshita, K.; Kurashige, J.; Ishimoto, T.; Iwatsuki, M.; Watanabe, M.; Baba, H. Clinical impact of serum exosomal microRNA-21 as a clinical biomarker in human esophageal squamous cell carcinoma. Cancer 2013, 119, 1159–1167. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Liu, D.; Lai, W.W.; Wang, Y.C.; Loh, Y.P. Carboxypeptidase E mRNA: Overexpression predicts recurrence and death in lung adenocarcinoma cancer patients. Cancer Biomark. 2021, 1–9. [Google Scholar] [CrossRef]
- Fan, S.; Gao, X.; Chen, P.; Li, X. Carboxypeptidase E-DeltaN promotes migration, invasiveness, and epithelial-mesenchymal transition of human osteosarcoma cells via the Wnt-beta-catenin pathway. Biochem. Cell Biol. 2019, 97, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.R.K.; Dupart, E.; Al-Sweel, N.; Chen, A.; Cawley, N.X.; Loh, Y.P. Carboxypeptidase E promotes cancer cell survival, but inhibits migration and invasion. Cancer Lett. 2013, 341, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schafer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, J.A. Cycling to cancer with cyclin D1. Cancer Biol. Ther. 2002, 1, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Shintani, M.; Okazaki, A.; Masuda, T.; Kawada, M.; Ishizuka, M.; Doki, Y.; Weinstein, I.B.; Imoto, M. Overexpression of cyclin DI contributes to malignant properties of esophageal tumor cells by increasing VEGF production and decreasing Fas expression. Anticancer Res. 2002, 22, 639–647. [Google Scholar]
- Zheng, K.; Cubero, F.J.; Nevzorova, Y.A. c-MYC-Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes 2017, 8, 123. [Google Scholar] [CrossRef]
- Akita, H.; Marquardt, J.U.; Durkin, M.E.; Kitade, M.; Seo, D.; Conner, E.A.; Andersen, J.B.; Factor, V.M.; Thorgeirsson, S.S. MYC activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer Res. 2014, 74, 5903–5913. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.K.; Yang, X.; Kim, S.K.; Mafi, A.; Saiz-Sanchez, D.; Villanueva-Anguita, P.; Xiao, L.; Inoue, A.; Goddard, W.A., 3rd; Loh, Y.P. Novel interaction between neurotrophic factor-alpha1/carboxypeptidase E and serotonin receptor, 5-HTR1E, protects human neurons against oxidative/neuroexcitotoxic stress via beta-arrestin/ERK signaling. Cell. Mol. Life Sci. 2021, 79, 24. [Google Scholar] [CrossRef]
- Bai, Z.; Feng, M.; Du, Y.; Cong, L.; Cheng, Y. Carboxypeptidase E down-regulation regulates transcriptional and epigenetic profiles in pancreatic cancer cell line: A network analysis. Cancer Biomark. 2020, 29, 79–88. [Google Scholar] [CrossRef]
- Skalka, N.; Caspi, M.; Lahav-Ariel, L.; Loh, Y.P.; Hirschberg, K.; Rosin-Arbesfeld, R. Carboxypeptidase E (CPE) inhibits the secretion and activity of Wnt3a. Oncogene 2016, 35, 6416–6428. [Google Scholar] [CrossRef]
- Xiao, L.; Yang, X.; Sharma, V.K.; Loh, Y.P. Cloning, gene regulation, and neuronal proliferation functions of novel N-terminal-truncated carboxypeptidase E/neurotrophic factor-alphal variants in embryonic mouse brain. FASEB J. 2019, 33, 808–820. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hareendran, S.; Albraidy, B.; Yang, X.; Liu, A.; Breggia, A.; Chen, C.C.; Loh, Y.P. Exosomal Carboxypeptidase E (CPE) and CPE-shRNA-Loaded Exosomes Regulate Metastatic Phenotype of Tumor Cells. Int. J. Mol. Sci. 2022, 23, 3113. https://doi.org/10.3390/ijms23063113
Hareendran S, Albraidy B, Yang X, Liu A, Breggia A, Chen CC, Loh YP. Exosomal Carboxypeptidase E (CPE) and CPE-shRNA-Loaded Exosomes Regulate Metastatic Phenotype of Tumor Cells. International Journal of Molecular Sciences. 2022; 23(6):3113. https://doi.org/10.3390/ijms23063113
Chicago/Turabian StyleHareendran, Sangeetha, Bassam Albraidy, Xuyu Yang, Aiyi Liu, Anne Breggia, Clark C. Chen, and Y. Peng Loh. 2022. "Exosomal Carboxypeptidase E (CPE) and CPE-shRNA-Loaded Exosomes Regulate Metastatic Phenotype of Tumor Cells" International Journal of Molecular Sciences 23, no. 6: 3113. https://doi.org/10.3390/ijms23063113
APA StyleHareendran, S., Albraidy, B., Yang, X., Liu, A., Breggia, A., Chen, C. C., & Loh, Y. P. (2022). Exosomal Carboxypeptidase E (CPE) and CPE-shRNA-Loaded Exosomes Regulate Metastatic Phenotype of Tumor Cells. International Journal of Molecular Sciences, 23(6), 3113. https://doi.org/10.3390/ijms23063113