
Crosstalk between p38 MAPK and GR Signaling


{kind=link}
Abstract
1. Introduction
2. Crosstalk between p38 and GR
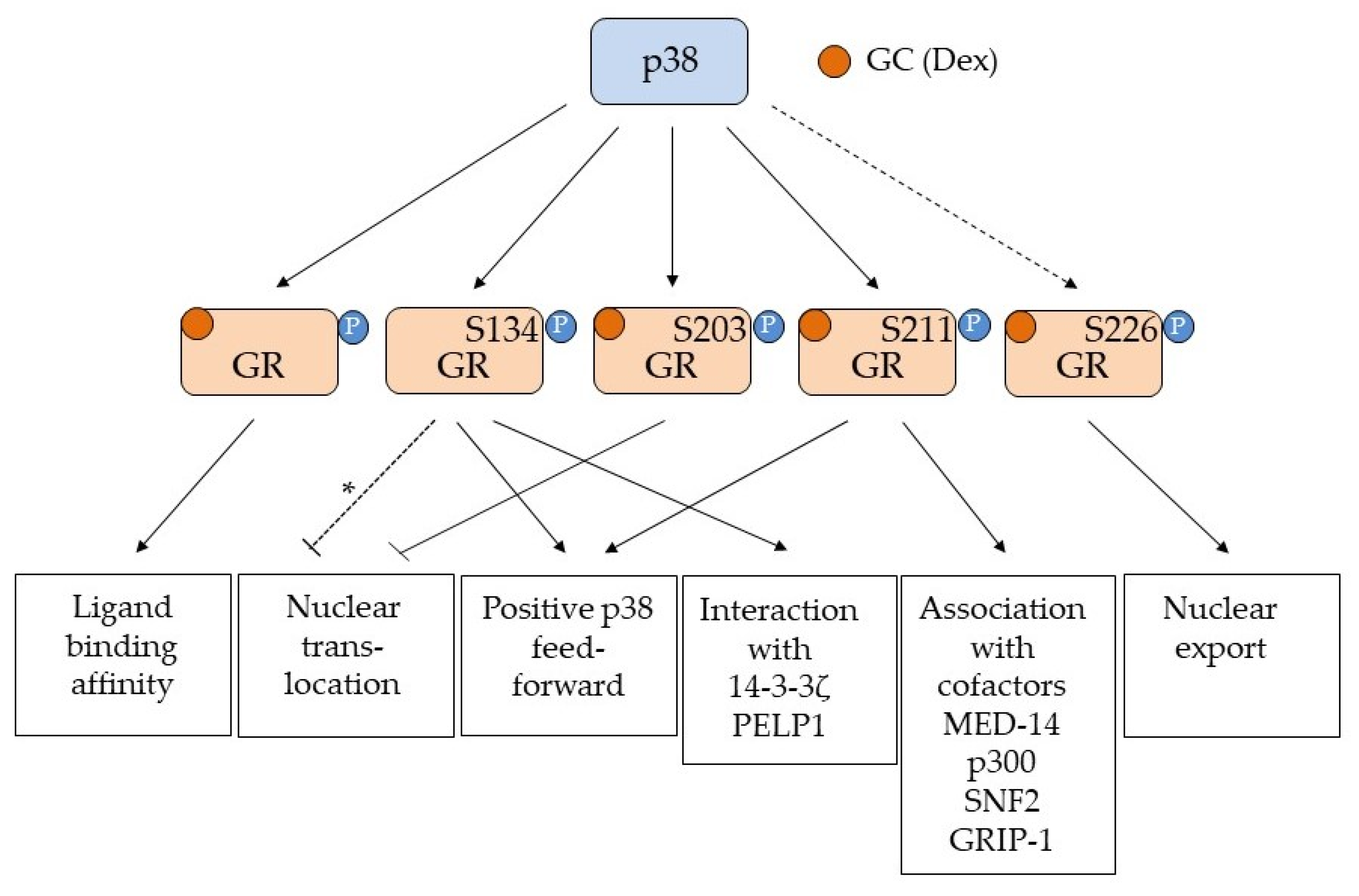
2.1. p38-Directed GR Phosphorylation and Regulation
2.2. GR Regulated p38 MAPK Phosphorylation and Dephosphorylation
2.3. Clinical Aspects of p38 and GR Crosstalk
2.4. Cancer-Related Interactions
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Argüelles, S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38γ and p38δ: From Spectators to Key Physiological Players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Gu, Z.; Li, L.; Liu, Y.; Wang, L.; Su, L. p38 MAPK-MK2 pathway regulates the heat-stress-induced accumulation of reactive oxygen species that mediates apoptotic cell death in glial cells. Oncol. Lett. 2017, 15, 775–782. [Google Scholar] [CrossRef]
- Wang, J.; Wang, G.; Cheng, D.; Huang, S.; Chang, A.; Tan, X.; Wang, Q.; Zhao, S.; Wu, D.; Liu, A.T.; et al. Her2 promotes early dissemination of breast cancer by suppressing the p38-MK2-Hsp27 pathway that is targetable by Wip1 inhibition. Oncogene 2020, 39, 6313–6326. [Google Scholar] [CrossRef]
- Yong, H.-Y.; Koh, M.-S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef]
- Hölscher, C.; Gräb, J.; Hölscher, A.; Müller, A.L.; Schäfer, S.C.; Rybniker, J. Chemical p38 MAP kinase inhibition constrains tissue inflammation and improves antibiotic activity in Mycobacterium tuberculosis-infected mice. Sci. Rep. 2020, 10, 13629. [Google Scholar] [CrossRef]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Vockley, C.M.; D’Ippolito, A.M.; McDowell, I.C.; Majoros, W.H.; Safi, A.; Song, L.; Crawford, G.E.; Reddy, T.E. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 2016, 166, 1269–1281.e19. [Google Scholar] [CrossRef] [PubMed]
- Thormann, V.; Rothkegel, M.C.; Schöpflin, R.; Glaser, L.V.; Djuric, P.; Li, N.; Chung, H.R.; Schwahn, K.; Vingron, M.; Meijsing, S.H. Genomic dissection of enhancers uncovers principles of combinatorial regulation and cell type-specific wiring of enhancer-promoter contacts. Nucleic Acids Res. 2018, 46, 2868–2882. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006, 16, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J. Biol. Chem. 1996, 271, 9550–9559. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Ramírez, P.; Tliba, O. Glucocorticoid Receptor β (GRβ): Beyond Its Dominant-Negative Function. Int. J. Mol. Sci. 2021, 22, 3649. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Sellner, S.; Kocabey, S.; Zhang, T.; Nekolla, K.; Hutten, S.; Krombach, F.; Liedl, T.; Rehberg, M. Dexamethasone-conjugated DNA nanotubes as anti-inflammatory agents in vivo. Biomaterials 2017, 134, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Spokoini, R.; Kfir-Erenfeld, S.; Cohen, O.; Yefenof, E. Chapter 6 Mechanisms Regulating the Susceptibility of Hematopoietic Malignancies to Glucocorticoid-Induced Apoptosis. Adv. Cancer Res. 2008, 101, 127–248. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Development of New Drugs for COPD. Curr. Med. Chem. 2013, 20, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Jiménez-Panizo, A.; Alegre-Martí, A.; Estébanez-Perpiñá, E.; Caelles, C.; Pérez, P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. Int. J. Mol. Sci. 2021, 22, 10049. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.R.; Lane, S.J.; Cidlowski, J.A.; Staynov, D.Z.; Lee, T.H. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor β-isoform. J. Allergy Clin. Immunol. 2000, 105, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tao, N.; Zheng, L.; Sun, T. LL-37 restored glucocorticoid sensitivity impaired by virus dsRNA in lung. Int. Immunopharmacol. 2020, 79, 106057. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.; Caiazzo, E.; McSharry, C.; Guzik, T.J.; Maffia, P. Why do some asthma patients respond poorly to glucocorticoid therapy? Pharmacol. Res. 2020, 160, 105189. [Google Scholar] [CrossRef] [PubMed]
- Rider, L.; Hirasawa, N.; Santini, F.; Beaven, M.A. Activation of the mitogen-activated protein kinase cascade is suppressed by low concentrations of dexamethasone in mast cells. J. Immunol. 1996, 157, 2374–2380. [Google Scholar] [PubMed]
- Krstic, M.D.; Rogatsky, I.; Yamamoto, K.R.; Garabedian, M.J. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol. Cell. Biol. 1997, 17, 3947–3954. [Google Scholar] [CrossRef] [PubMed]
- Swantek, J.L.; Cobb, M.H.; Geppert, T.D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: Glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol. Cell. Biol. 1997, 17, 6274–6282. [Google Scholar] [CrossRef] [PubMed]
- Lasa, M.; Brook, M.; Saklatvala, J.; Clark, A.R. Dexamethasone Destabilizes Cyclooxygenase 2 mRNA by Inhibiting Mitogen-Activated Protein Kinase p38. Mol. Cell. Biol. 2001, 21, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I. p38 Mitogen-activated protein kinase–induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Frederick, J.; Garabedian, M.J. Deciphering the Phosphorylation “Code” of the Glucocorticoid Receptor In Vivo. J. Biol. Chem. 2002, 277, 26573–26580. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.; Kumar, R.; Thompson, E.B. p38 Mitogen-Activated Protein Kinase (MAPK) Is a Key Mediator in Glucocorticoid-Induced Apoptosis of Lymphoid Cells: Correlation between p38 MAPK Activation and Site-Specific Phosphorylation of the Human Glucocorticoid Receptor at Serine 211. Mol. Endocrinol. 2005, 19, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Ng, S.S.M.; Lambrou, G.; Pervanidou, P.; Wang, Y.; Chrousos, G.P.; Kino, T. AMPK Regulates Metabolic Actions of Glucocorticoids by Phosphorylating the Glucocorticoid Receptor through p38 MAPK. Mol. Endocrinol. 2010, 24, 1748–1764. [Google Scholar] [CrossRef] [PubMed]
- Bouazza, B.; Debba-Pavard, M.; Amrani, Y.; Isaacs, L.; O’Connell, D.; Ahamed, S.; Formella, D.; Tliba, O. Basal p38 mitogen-activated protein kinase regulates unliganded glucocorticoid receptor function in airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 301–315. [Google Scholar] [PubMed]
- Itoh, M.; Adachi, M.; Yasui, H.; Takekawa, M.; Tanaka, H.; Imai, K. Nuclear Export of Glucocorticoid Receptor is Enhanced by c-Jun N-Terminal Kinase-Mediated Phosphorylation. Mol. Endocrinol. 2002, 16, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; To, Y.; Kobayashi, Y.; Adcock, I.; Barnes, P.J.; Ito, K. p38 Mitogen-Activated Protein Kinase-γ Inhibition by Long-Acting β2 Adrenergic Agonists Reversed Steroid Insensitivity in Severe Asthma. Mol. Pharmacol. 2011, 80, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; Hakim, A.; Kobayashi, Y.; Meah, S.; Usmani, O.S.; Chung, K.F.; Barnes, P.J.; Ito, K. Restoration of Corticosteroid Sensitivity by p38 Mitogen Activated Protein Kinase Inhibition in Peripheral Blood Mononuclear Cells from Severe Asthma. PLoS ONE 2012, 7, e41582. [Google Scholar] [CrossRef] [PubMed]
- Lea, S.; Li, J.; Plumb, J.; Gaffey, K.; Mason, S.; Gaskell, R.; Harbron, C.; Singh, D. P38 MAPK and glucocorticoid receptor crosstalk in bronchial epithelial cells. Klin. Wochenschr. 2020, 98, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Galliher-Beckley, A.J.; Williams, J.G.; Cidlowski, J. Ligand-Independent Phosphorylation of the Glucocorticoid Receptor Integrates Cellular Stress Pathways with Nuclear Receptor Signaling. Mol. Cell. Biol. 2011, 31, 4663–4675. [Google Scholar] [CrossRef] [PubMed]
- Regan Anderson, T.M.; Ma, S.H.; Raj, G.V.; Cidlowski, J.A.; Helle, T.M.; Knutson, T.P.; Krutilina, R.I.; Seagroves, T.N.; Lange, C.A. Breast Tumor Kinase (Brk/PTK6) Is Induced by HIF, Glucocorticoid Receptor, and PELP1-Mediated Stress Signaling in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Kerkvliet, C.P.; Dwyer, A.R.; Diep, C.H.; Oakley, R.H.; Liddle, C.; Cidlowski, J.A.; Lange, C.A. Glucocorticoid receptors are required effectors of TGFβ1-induced p38 MAPK signaling to advanced cancer phenotypes in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; Da Silva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct Reversal of Glucocorticoid Resistance by AKT Inhibition in Acute Lymphoblastic Leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Habib, T.; Sadoun, A.; Nader, N.; Suzuki, S.; Liu, W.; Jithesh, P.V.; Kino, T. AKT1 has dual actions on the glucocorticoid receptor by cooperating with 14-3-3. Mol. Cell. Endocrinol. 2017, 439, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Souvatzoglou, E.; De Martino, M.U.; Tsopanomihalu, M.; Wan, Y.; Chrousos, G.P. Protein 14-3-3sigma interacts with and favors cytoplasmic subcellular localization of the glucocorticoid receptor, acting as a negative regulator of the glucocorticoid signaling pathway. J. Biol. Chem. 2003, 278, 25651–25656. [Google Scholar] [CrossRef] [PubMed]
- Basta-Kaim, A.; Budziszewska, B.; Jaworska-Feil, L.; Tetich, M.; Kubera, M.; Zajicova, A.; Holan, V.; Lasoń, W. Effects of lipopolysaccharide and chlorpromazine on glucocorticoid receptor-mediated gene transcription and immunoreactivity: A possible involvement of p38-MAP kinase. Eur. Neuropsychopharmacol. 2004, 14, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, H.; Miller, A.H. Interleukin 1α (IL-1α) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol. Psychiatry 2003, 9, 65–75. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blind, R.D.; Garabedian, M.J. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J. Steroid Biochem. Mol. Biol. 2008, 109, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dang, T.; Blind, R.D.; Wang, Z.; Cavasotto, C.N.; Hittelman, A.B.; Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Glucocorticoid Receptor Phosphorylation Differentially Affects Target Gene Expression. Mol. Endocrinol. 2008, 22, 1754–1766. [Google Scholar] [CrossRef]
- Khan, S.H.; McLaughlin, W.A.; Kumar, R. Site-specific phosphorylation regulates the structure and function of an intrinsically disordered domain of the glucocorticoid receptor. Sci. Rep. 2017, 7, 15440. [Google Scholar] [CrossRef] [PubMed]
- Avenant, C.; Kotitschke, A.; Hapgood, J.P. Glucocorticoid Receptor Phosphorylation Modulates Transcription Efficacy through GRIP-1 Recruitment. Biochemistry 2010, 49, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Szatmáry, Z.; Garabedian, M.J.; Vilček, J. Inhibition of Glucocorticoid Receptor-mediated Transcriptional Activation by p38 Mitogen-activated Protein (MAP) Kinase. J. Biol. Chem. 2004, 279, 43708–43715. [Google Scholar] [CrossRef] [PubMed]
- Beck, I.; Berghe, W.V.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef] [PubMed]
- Ayroldi, E.; Cannarile, L.; Migliorati, G.; Nocentini, G.; Delfino, D.V.; Riccardi, C. Mechanisms of the anti-inflammatory effects of glucocorticoids: Genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012, 26, 4805–4820. [Google Scholar] [CrossRef] [PubMed]
- Kassel, O.; Sancono, A.; Krätzschmar, J.; Kreft, B.; Stassen, M.; Cato, A.C. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001, 20, 7108–7116. [Google Scholar] [CrossRef] [PubMed]
- Seternes, O.-M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Brown, D.E.; Brewer, J.A.; Vogt, S.K.; Muglia, L.J. Macrophage glucocorticoid receptors regulate Toll-like receptor 4–mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 2007, 109, 4313–4319. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.P.; Saklatvala, J.; Clark, A.R. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qiu, J.; Wang, J.; Zhong, Y.; Zhu, J.; Chen, Y. Corticosterone-induced rapid phosphorylation of p38 and JNK mitogen-activated protein kinases in PC12 cells. FEBS Lett. 2001, 492, 210–214. [Google Scholar] [CrossRef]
- Qi, A.-Q.; Qiu, J.; Xiao, L.; Chen, Y.-Z. Rapid activation of JNK and p38 by glucocorticoids in primary cultured hippocampal cells. J. Neurosci. Res. 2005, 80, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Arthurton, L.; Akujuru, E.; Pearson, T.; Steverding, D.; Protasi, F.; Mutungi, G. Membrane glucocorticoid receptors are localised in the extracellular matrix and signal through the MAPK pathway in mammalian skeletal muscle fibres. J. Physiol. 2015, 593, 2679–2692. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, G.Q.; Peng, J.P.; Shen, C. Glucocorticoids induce apoptosis and matrix metalloproteinase-13 expression in chondrocytes through the NOX4/ROS/p38 MAPK pathway. J. Steroid Biochem. Mol. Biol. 2018, 181, 52–62. [Google Scholar] [CrossRef]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, P.K.; Khorasani, N.; Baker, J.; Johnson, M.; Chung, K.F. Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 283–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higham, A.; Singh, D. Dexamethasone and p38 MAPK inhibition of cytokine production from human lung fibroblasts. Fundam. Clin. Pharmacol. 2021, 35, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Rebeyrol, C.; Saint-Criq, V.; Guillot, L.; Riffault, L.; Corvol, H.; Chadelat, K.; Ray, D.; Clement, A.; Tabary, O.; Le Rouzic, P. Glucocorticoids reduce inflammation in cystic fibrosis bronchial epithelial cells. Cell. Signal. 2012, 24, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Xu, R.; Li, M.; Zhao, Q.; Ren, X.; Li, Z.; Cao, J.; Zang, W. Glucocorticoid receptor inhibit the activity of NF-κB through p38 signaling pathway in spinal cord in the spared nerve injury rats. Life Sci. 2018, 208, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Yubero, S.; Ramudo, L.; Manso, M.; De Dios, I. Mechanisms of dexamethasone-mediated chemokine down-regulation in mild and severe acute pancreatitis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2009, 1792, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Bläuer, M.; Sand, J.; Laukkarinen, J. Regulation of p38 MAPK and glucocorticoid receptor activation by hydrocortisone in mono-and co-cultured pancreatic acinar and stellate cells. Pancreatology 2021, 21, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Roila, F.; Fatigoni, S. New antiemetic drugs. Ann. Oncol. 2006, 17, ii96–ii100. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, P.J.; Kris, M.G.; Basch, E.; Bohlke, K.; Barbour, S.Y.; Clark-Snow, R.A.; Danso, M.A.; Dennis, K.; Dupuis, L.L.; Dusetzina, S.B.; et al. Antiemetics: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 2782–2797. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ge, J.; Li, Q.; Guo, X.; Gu, L.; Ma, Z.-G.; Li, X.-H.; Zhu, Y.-P. Low-dose anisomycin sensitizes glucocorticoid-resistant T-acute lymphoblastic leukemia CEM-C1 cells to dexamethasone-induced apoptosis through activation of glucocorticoid receptor and p38-MAPK/JNK. Leuk. Lymphoma 2014, 55, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.D. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 2008, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.; Harkless, R.V.; Oh, A.; Bowie, K.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid receptor modulation decreases ER-positive breast cancer cell proliferation and suppresses wild-type and mutant ER chromatin association. Breast Cancer Res. BCR 2019, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Mu, D.-L.; Jin, K.-M.; Li, X.-Y.; Wang, D.-X. Perioperative Glucocorticoids are Associated with Improved Recurrence-Free Survival After Pancreatic Cancer Surgery: A Retrospective Cohort Study with Propensity Score-Matching. Ther. Clin. Risk Manag. 2021, 17, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, H.T.; Mellemkjaer, L.; Nielsen, G.L.; Baron, J.A.; Olsen, J.H.; Karagas, M.R. Skin Cancers and Non-Hodgkin Lymphoma Among Users of Systemic Glucocorticoids: A Population-Based Cohort Study. JNCI J. Natl. Cancer Inst. 2004, 96, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, M.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Muenst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, G.; Zhang, H.; Xiong, Q.; Cheng, F.; Wang, H.; Luo, J.; Zhang, Y.; Shi, P.; Xu, J.; et al. Dexamethasone enhances the lung metastasis of breast cancer via a PI3K-SGK1-CTGF pathway. Oncogene 2021, 40, 5367–5378. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Aleksandrowicz, E.; Schönsiegel, F.; Gröner, D.; Bauer, N.; Nwaeburu, C.C.; Zhao, Z.; Gladkich, J.; Hoppe-Tichy, T.; Yefenof, E.; et al. Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFβ and JNK/AP-1. Cell Death Dis. 2017, 8, e3064. [Google Scholar] [CrossRef] [PubMed]
- Regan Anderson, T.M.; Ma, S.; Perez Kerkvliet, C.; Peng, Y.; Helle, T.M.; Krutilina, R.I.; Raj, G.V.; Cidlowski, J.A.; Ostrander, J.H.; Schwertfeger, K.L.; et al. Taxol Induces Brk-dependent Prosurvival Phenotypes in TNBC Cells through an AhR/GR/HIF-driven Signaling Axis. Mol. Cancer Res. MCR 2018, 16, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Mattern, J.; Büchler, M.W.; Herr, I. Cell Cycle Arrest by Glucocorticoids May Protect Normal Tissue and Solid Tumors from Cancer Therapy. Cancer Biol. Ther. 2007, 6, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar] [PubMed]
- de Olano, N.; Koo, C.Y.; Monteiro, L.J.; Pinto, P.H.; Gomes, A.R.; Aligue, R.; Lam, E.W.-F. The p38 MAPK–MK2 Axis Regulates E2F1 and FOXM1 Expression after Epirubicin Treatment. Mol. Cancer Res. 2012, 10, 1189–1202. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeyen, L.; Seternes, O.M.; Mikkola, I. Crosstalk between p38 MAPK and GR Signaling. Int. J. Mol. Sci. 2022, 23, 3322. https://doi.org/10.3390/ijms23063322
Zeyen L, Seternes OM, Mikkola I. Crosstalk between p38 MAPK and GR Signaling. International Journal of Molecular Sciences. 2022; 23(6):3322. https://doi.org/10.3390/ijms23063322
Chicago/Turabian StyleZeyen, Lisa, Ole Morten Seternes, and Ingvild Mikkola. 2022. "Crosstalk between p38 MAPK and GR Signaling" International Journal of Molecular Sciences 23, no. 6: 3322. https://doi.org/10.3390/ijms23063322
APA StyleZeyen, L., Seternes, O. M., & Mikkola, I. (2022). Crosstalk between p38 MAPK and GR Signaling. International Journal of Molecular Sciences, 23(6), 3322. https://doi.org/10.3390/ijms23063322