
Characterization of Chromosomal Breakpoints in 12 Cases with 8p Rearrangements Defines a Continuum of Fragility of the Region

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
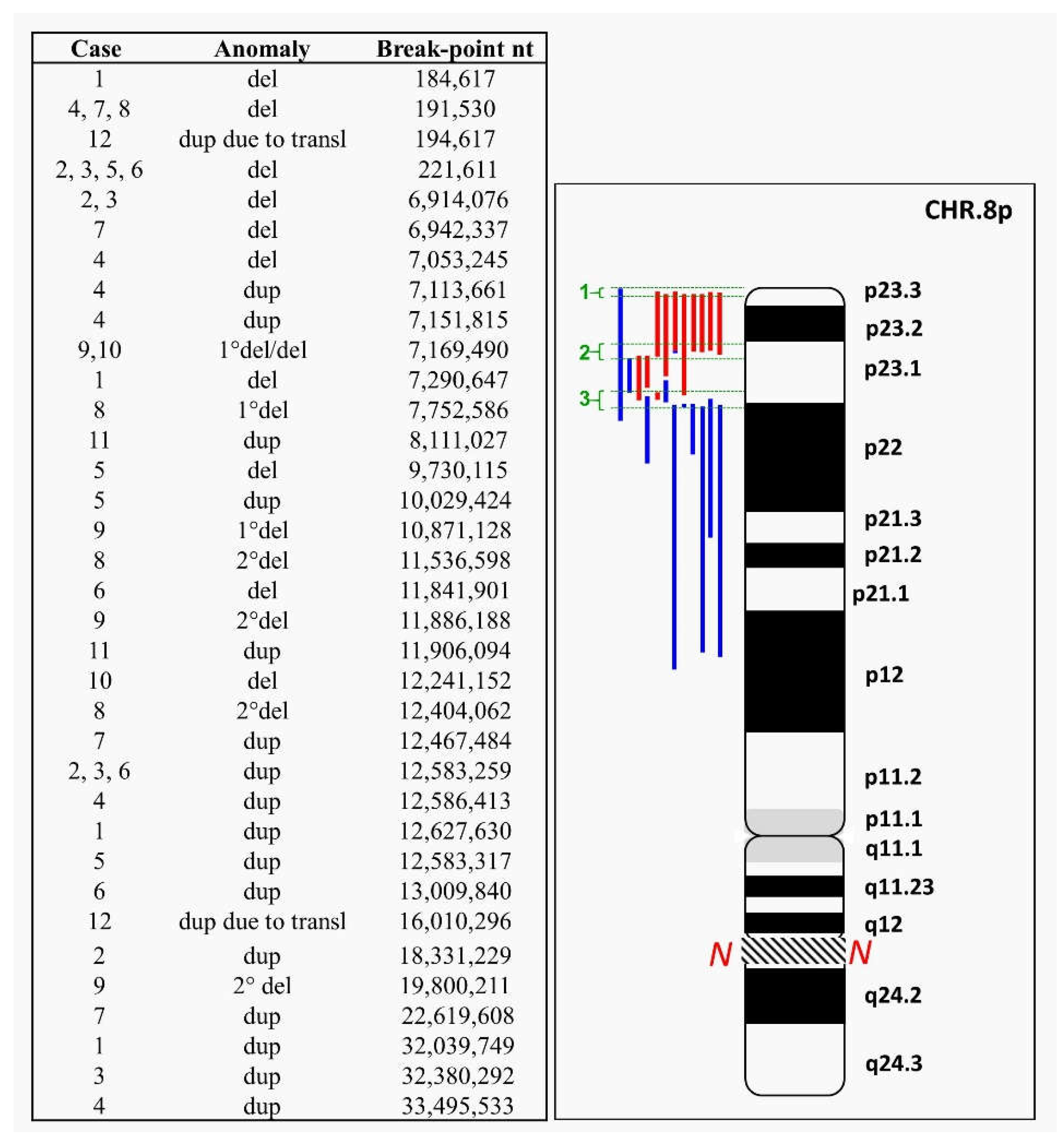
2. Case Reports and Cytogenetics—Genomics Description
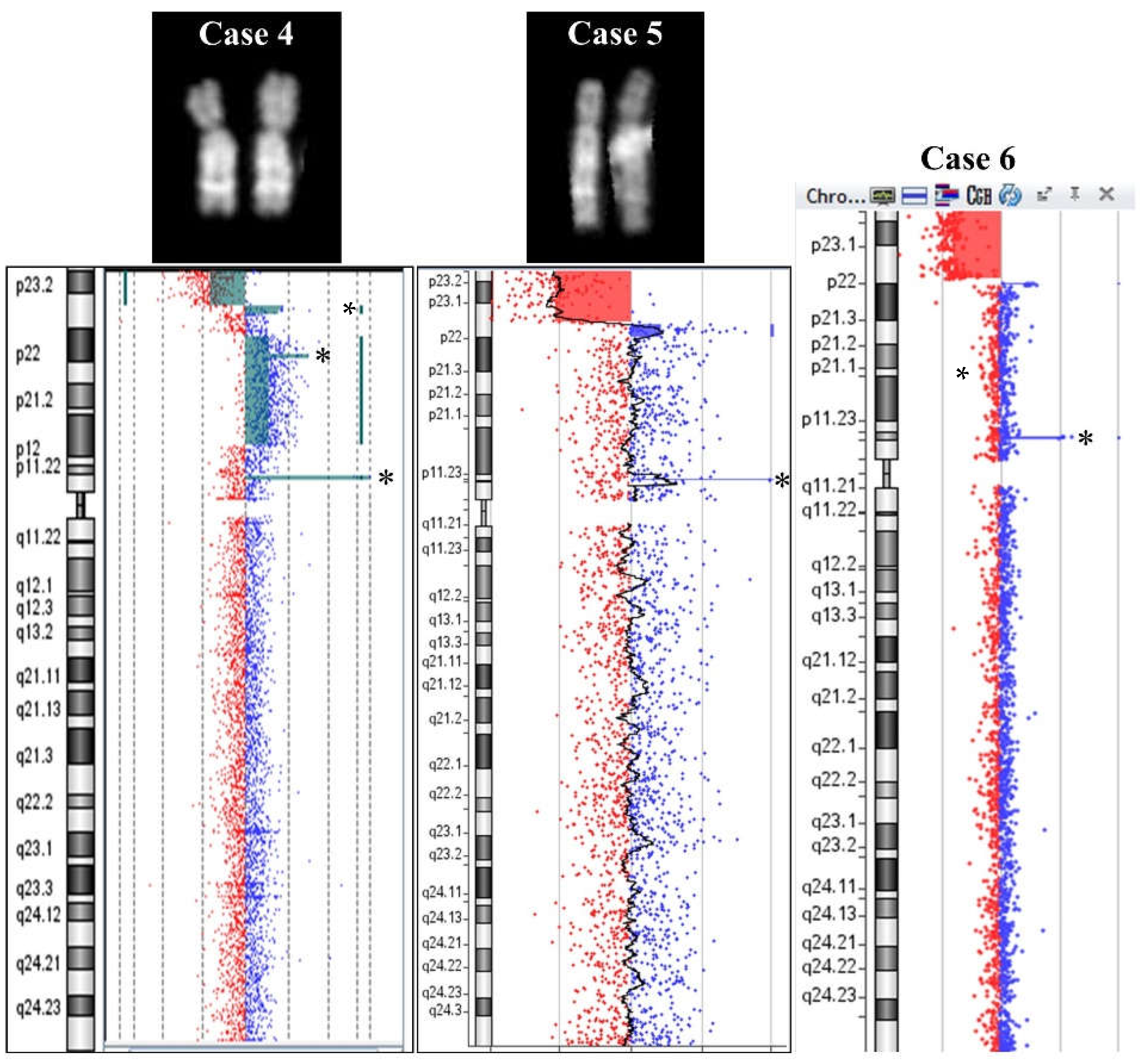
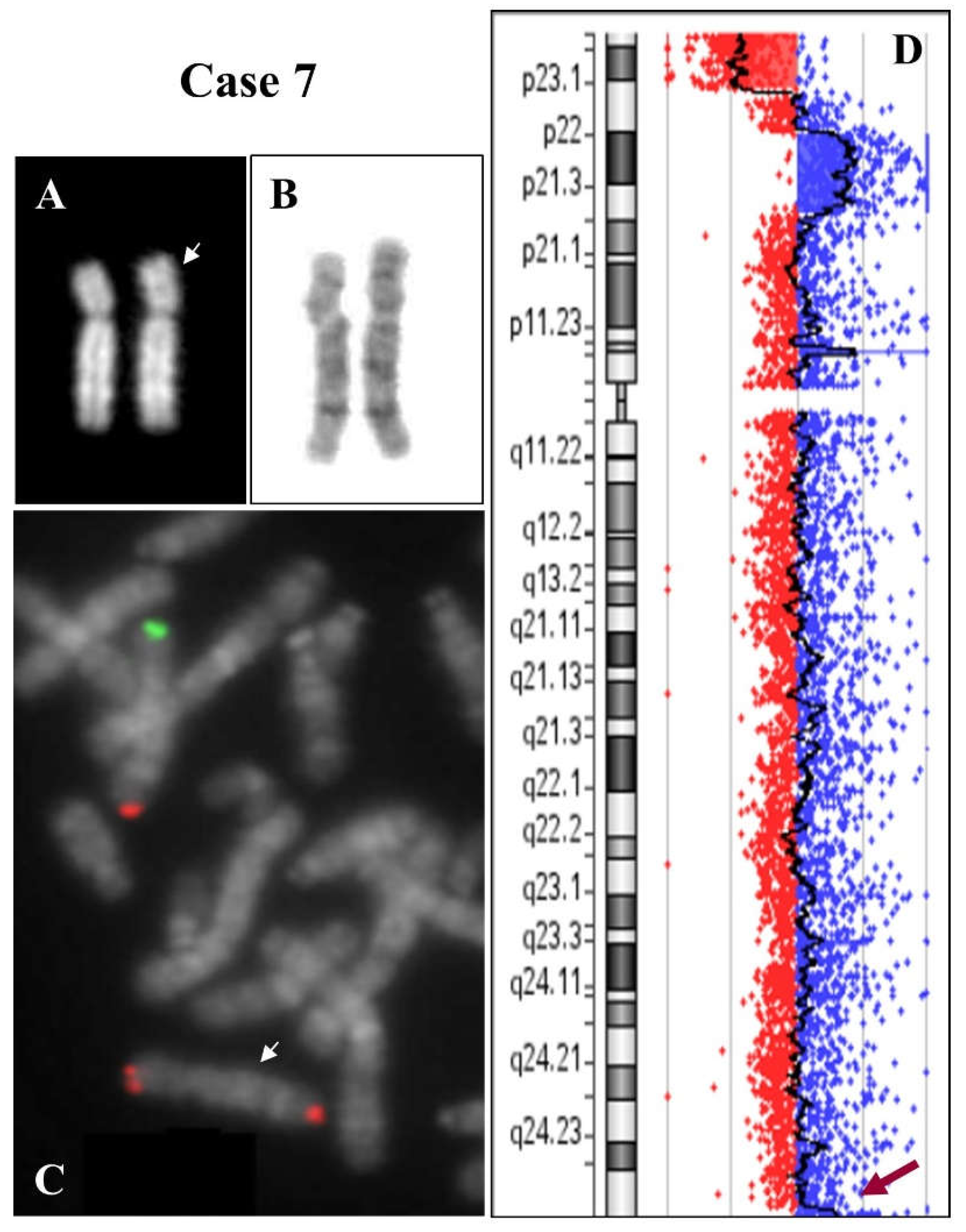
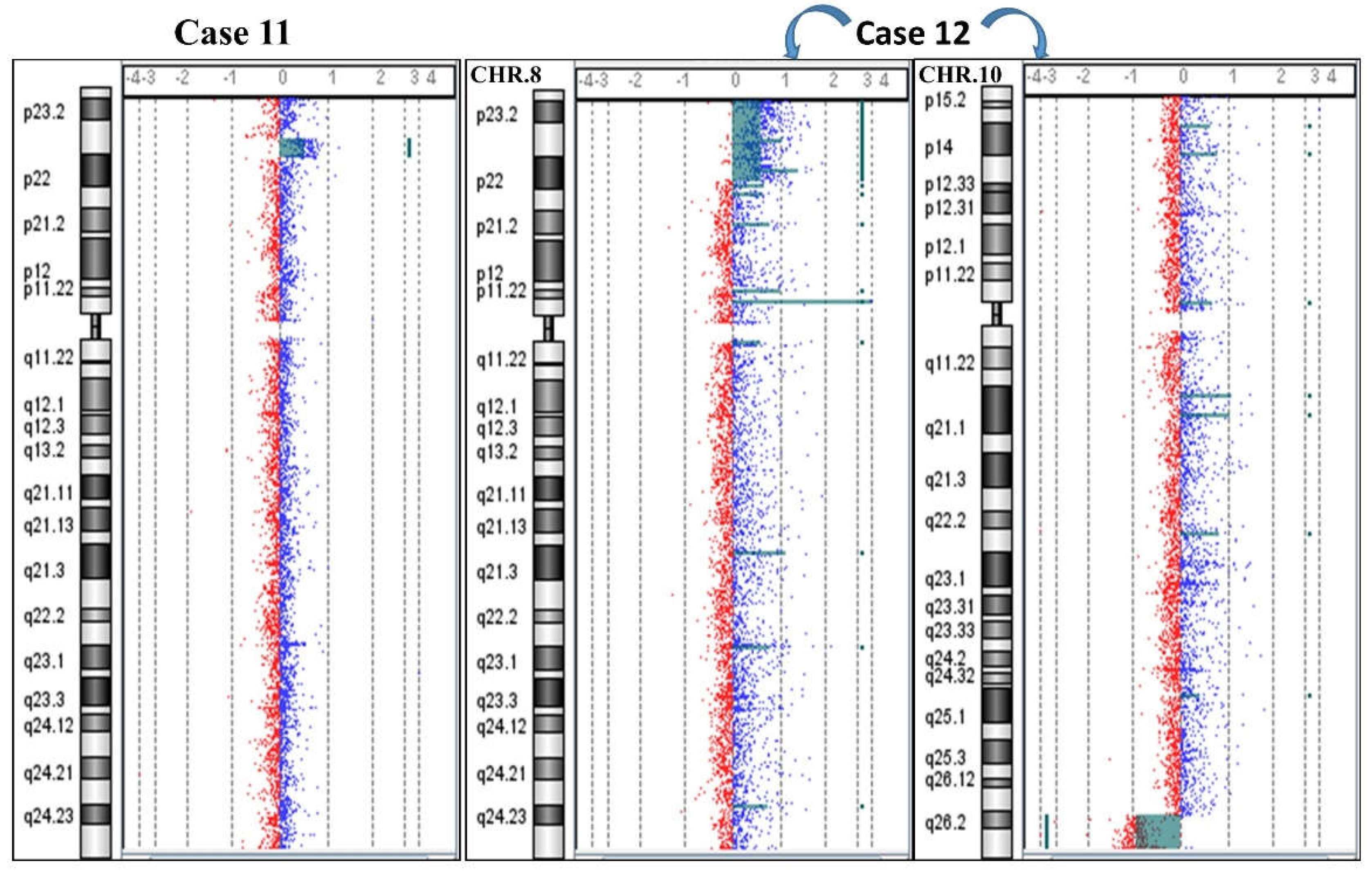
2.1. 8p Inverted Duplication with Deletion
2.2. Isolated Deletion or Duplication of 8p
2.3. Benign CNVs on 8p
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Chromosome Analysis
4.3. FISH Analysis
4.4. Array Comparative Genomic Hybridization (Array CGH)
4.5. UCSC Genome Browser Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Floridia, G.; Piantanida, M.; Minelli, A.; Dellavecchia, C.; Bonaglia, C.; Rossi, E.; Gimelli, G.; Croci, G.; Franchi, F.; Gilgenkrantz, S.; et al. The same molecular mechanism at the maternal meiosis I produces mono- and dicentric 8p duplications. Am. J. Hum. Genet. 1996, 58, 785–796. [Google Scholar] [PubMed]
- Giglio, S.; Broman, K.W.; Matsumoto, N.; Calvari, V.; Gimelli, G.; Neumann, T.; Ohashi, H.; Voullaire, L.; Larizza, D.; Giorda, R.; et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am. J. Hum. Genet. 2001, 68, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, O.; Kurosawa, K.; Ida, T.; Harada, N.; Kondoh, T.; Miyake, N.; Yoshiura, K.; Kishino, T.; Ohta, T.; Niikawa, N.; et al. Molecular characterization of inv dup del(8p): Analysis of five cases. Am. J. Med. Genet. A 2004, 128, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Feldman, G.L.; Weiss, L.; Phelan, M.C.; Schroer, R.J.; Van Dyke, D.L. Inverted duplication of 8p: Ten new patients and review of the literature. Am. J. Med. Genet. 1993, 47, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Carey, J.C.; South, S.T. Wolf-Hirschhorn syndrome: A review and update. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 216–223. [Google Scholar] [CrossRef]
- Nevado, J.; Ho, K.S.; Zollino, M.; Blanco, R.; Cobaleda, C.; Golzio, C.; Beaudry-Bellefeuille, I.; Berrocoso, S.; Limeres, J.; Barrúz, P.; et al. International meeting on Wolf-Hirschhorn syndrome: Update on the nosology and new insights on the pathogenic mechanisms for seizures and growth delay. Am. J. Med. Genet. A 2020, 182, 257–267. [Google Scholar] [CrossRef]
- Giglio, S.; Calvari, V.; Gregato, G.; Gimelli, G.; Camanini, S.; Giorda, R.; Ragusa, A.; Guerneri, S.; Selicorni, A.; Stumm, M.; et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am. J. Hum. Genet. 2002, 71, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Zollino, M.; Murdolo, M.; Marangi, G.; Pecile, V.; Galasso, C.; Mazzanti, L.; Neri, G. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: Genotype-phenotype correlation analysis of 80 patients and literature review. Am. J. Med. Genet. C Semin. Med. Genet. 2008, 148, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Glusman, G.; Yanai, I.; Rubin, I.; Lancet, D. The complete human olfactory subgenome. Genome Res. 2001, 11, 685–702. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, R.; Wilson, M.; Voullaire, L. Distal 8p deletion (8p23.1----8pter): A common deletion? J. Med. Genet. 1992, 29, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Lupski, J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998, 14, 417–422. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002, 18, 74–82. [Google Scholar] [CrossRef]
- Hollox, E.J.; Barber, J.C.; Brookes, A.J.; Armour, J.A. Defensins and the dynamic genome: What we can learn from structural variation at human chromosome band 8p23.1. Genome Res. 2008, 18, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, G.A.; Vollger, M.R.; Hsieh, P.; Mao, Y.; Liskovykh, M.A.; Koren, S.; Nurk, S.; Mercuri, L.; Dishuck, P.C.; Rhie, A.; et al. The structure, function and evolution of a complete human chromosome 8. Nature 2021, 593, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Salm, M.P.; Horswell, S.D.; Hutchison, C.E.; Speedy, H.E.; Yang, X.; Liang, L.; Schadt, E.E.; Cookson, W.O.; Wierzbicki, A.S.; Naoumova, R.P.; et al. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 2012, 22, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohajeri, K.; Cantsilieris, S.; Huddleston, J.; Nelson, B.J.; Coe, B.P.; Campbell, C.D.; Baker, C.; Harshman, L.; Munson, K.M.; Kronenberg, Z.N.; et al. Interchromosomal core duplicons drive both evolutionary instability and disease susceptibility of the Chromosome 8p23.1 region. Genome Res. 2016, 26, 1453–1467. [Google Scholar] [CrossRef] [Green Version]
- Ballarati, L.; Cereda, A.; Caselli, R.; Selicorni, A.; Recalcati, M.P.; Maitz, S.; Finelli, P.; Larizza, L.; Giardino, D. Genotype-phenotype correlations in a new case of 8p23.1 deletion and review of the literature. Eur. J. Med. Genet. 2011, 54, 55–59. [Google Scholar] [CrossRef]
- Shi, S.; Lin, S.; Chen, B.; Zhou, Y. Isolated chromosome 8p23.2-pter deletion: Novel evidence for developmental delay, intellectual disability, microcephaly and neurobehavioral disorders. Mol. Med. Rep. 2017, 16, 6837–6845. [Google Scholar] [CrossRef] [Green Version]
- Catusi, I.; Garzo, M.; Capra, A.P.; Briuglia, S.; Baldo, C.; Canevini, M.P.; Cantone, R.; Elia, F.; Forzano, F.; Galesi, O.; et al. 8p23.2-pter Microdeletions: Seven New Cases Narrowing the Candidate Region and Review of the Literature. Genes 2021, 12, 652. [Google Scholar] [CrossRef]
- Barber, J.C.; Rosenfeld, J.A.; Foulds, N.; Laird, S.; Bateman, M.S.; Thomas, N.S.; Baker, S.; Maloney, V.K.; Anilkumar, A.; Smith, W.E.; et al. 8p23.1 duplication syndrome; common, confirmed, and novel features in six further patients. Am. J. Med. Genet. A 2013, 161, 487–500. [Google Scholar] [CrossRef]
- Barber, J.C.; Rosenfeld, J.A.; Graham, J.M.; Kramer, N.; Lachlan, K.L.; Bateman, M.S.; Collinson, M.N.; Stadheim, B.F.; Turner, C.L.; Gauthier, J.N.; et al. Inside the 8p23.1 duplication syndrome; eight microduplications of likely or uncertain clinical significance. Am. J. Med. Genet. A 2015, 167, 2052–2064. [Google Scholar] [CrossRef] [PubMed]
- Buysse, K.; Antonacci, F.; Callewaert, B.; Loeys, B.; Fränkel, U.; Siu, V.; Mortier, G.; Speleman, F.; Menten, B. Unusual 8p inverted duplication deletion with telomere capture from 8q. Eur. J. Med. Genet. 2009, 52, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Graf, W.D. Telomere capture as a frequent mechanism for stabilization of the terminal chromosomal deletion associated with inverted duplication. Cytogenet. Genome Res. 2010, 129, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Inagaki, H.; Miyai, S.; Suzuki, F.; Naru, Y.; Shinkai, Y.; Kato, A.; Kanyama, K.; Mizuno, S.; Muramatsu, Y.; et al. The involvement of U-type dicentric chromosomes in the formation of terminal deletions with or without adjacent inverted duplications. Hum. Genet. 2020, 139, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Kostiner, D.R.; Nguyen, H.; Cox, V.A.; Cotter, P.D. Stabilization of a terminal inversion duplication of 8p by telomere capture from 18q. Cytogenet. Genome Res. 2002, 98, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Yu, W.; Shaw, C.A.; Kashork, C.D.; Shaffer, L.G. Monosomy 1p36 breakpoint junctions suggest pre-meiotic breakage-fusion-bridge cycles are involved in generating terminal deletions. Hum. Mol. Genet. 2003, 12, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Gajecka, M.; Shaffer, L.G. Monosomy 1p36 breakpoints indicate repetitive DNA sequence elements may be involved in generating and/or stabilizing some terminal deletions. Chromosome Res. 2004, 12, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Wakui, K.; Gajecka, M.; Shaffer, L.G. Translocation breakpoint mapping and sequence analysis in three monosomy 1p36 subjects with der(1)t(1;1)(p36;q44) suggest mechanisms for telomere capture in stabilizing de novo terminal rearrangements. Hum. Genet. 2004, 114, 198–206. [Google Scholar] [CrossRef]
- Rowe, L.R.; Lee, J.Y.; Rector, L.; Kaminsky, E.B.; Brothman, A.R.; Martin, C.L.; South, S.T. U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J. Med. Genet. 2009, 46, 694–702. [Google Scholar] [CrossRef]
- Chai, H.; Ji, W.; Wen, J.; DiAdamo, A.; Grommisch, B.; Hu, Q.; Szekely, A.M.; Li, P. Ring chromosome formation by intra-strand repairing of subtelomeric double stand breaks and clinico-cytogenomic correlations for ring chromosome 9. Am. J. Med. Genet. A 2020, 182, 3023–3028. [Google Scholar] [CrossRef]
- Knijnenburg, J.; van Haeringen, A.; Hansson, K.B.; Lankester, A.; Smit, M.J.; Belfroid, R.D.; Bakker, E.; Rosenberg, C.; Tanke, H.J.; Szuhai, K. Ring chromosome formation as a novel escape mechanism in patients with inverted duplication and terminal deletion. Eur. J. Hum. Genet. 2007, 15, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Maitz, S.; Crosti, F.; Sala, E.; Villa, N.; Spaccini, L.; Selicorni, A.; Rigoldi, M.; Conconi, D.; Dalprà, L.; et al. Refining the Phenotype of Recurrent Rearrangements of Chromosome 16. Int. J. Mol. Sci. 2019, 20, 1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diagnostica Citogenetica, Consensus. Associazione Italiana di Citogenetica Medica. Analysis 1995, 8, 12–42. [Google Scholar]
- International Standing Committee on Human Cytogenomic Nomenclature. ISCN: An International System for Human Cytoge-nomic Nomenclature; Karger: Basel, Switzerland, 2020. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redaelli, S.; Conconi, D.; Sala, E.; Villa, N.; Crosti, F.; Roversi, G.; Catusi, I.; Valtorta, C.; Recalcati, M.P.; Dalprà, L.; et al. Characterization of Chromosomal Breakpoints in 12 Cases with 8p Rearrangements Defines a Continuum of Fragility of the Region. Int. J. Mol. Sci. 2022, 23, 3347. https://doi.org/10.3390/ijms23063347
Redaelli S, Conconi D, Sala E, Villa N, Crosti F, Roversi G, Catusi I, Valtorta C, Recalcati MP, Dalprà L, et al. Characterization of Chromosomal Breakpoints in 12 Cases with 8p Rearrangements Defines a Continuum of Fragility of the Region. International Journal of Molecular Sciences. 2022; 23(6):3347. https://doi.org/10.3390/ijms23063347
Chicago/Turabian StyleRedaelli, Serena, Donatella Conconi, Elena Sala, Nicoletta Villa, Francesca Crosti, Gaia Roversi, Ilaria Catusi, Chiara Valtorta, Maria Paola Recalcati, Leda Dalprà, and et al. 2022. "Characterization of Chromosomal Breakpoints in 12 Cases with 8p Rearrangements Defines a Continuum of Fragility of the Region" International Journal of Molecular Sciences 23, no. 6: 3347. https://doi.org/10.3390/ijms23063347
APA StyleRedaelli, S., Conconi, D., Sala, E., Villa, N., Crosti, F., Roversi, G., Catusi, I., Valtorta, C., Recalcati, M. P., Dalprà, L., Lavitrano, M., & Bentivegna, A. (2022). Characterization of Chromosomal Breakpoints in 12 Cases with 8p Rearrangements Defines a Continuum of Fragility of the Region. International Journal of Molecular Sciences, 23(6), 3347. https://doi.org/10.3390/ijms23063347