
Colocalization and Interaction Study of Neuronal JNK3, JIP1, and β-Arrestin2 Together with PSD95


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. JNK3 Colocalizes with PSD95/JIP1 in Brain Cells
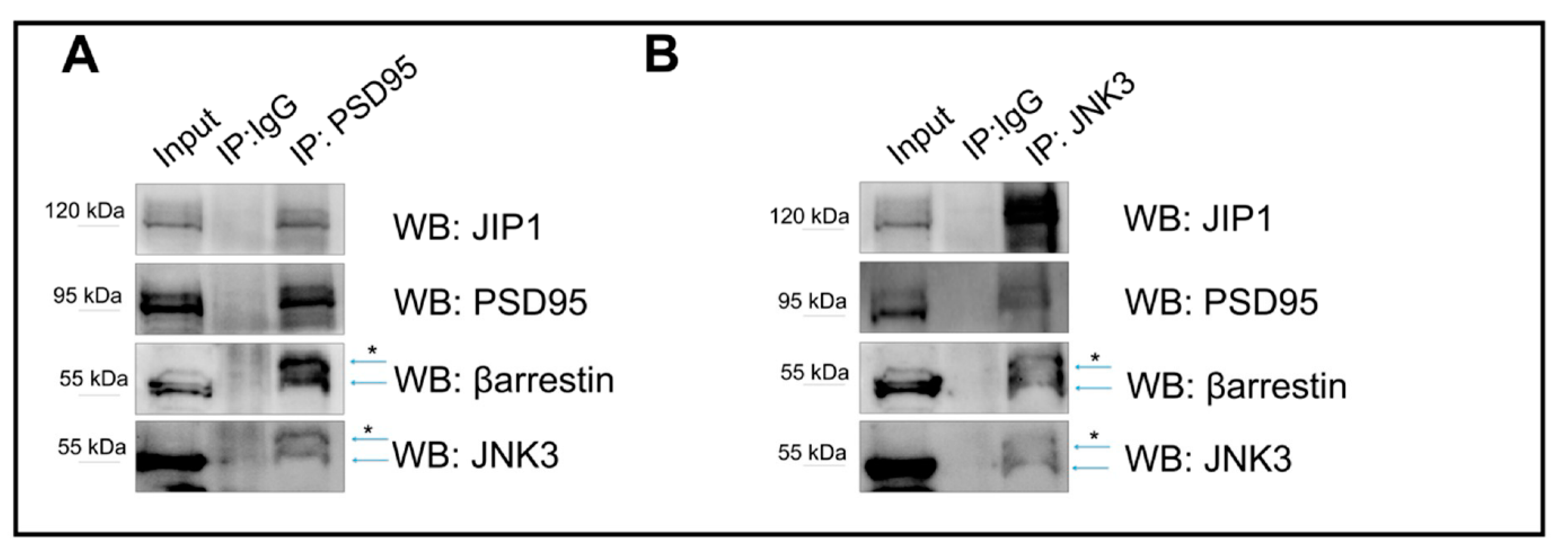
2.2. JNK3 Colocalizes and Forms a Complex with PSD95/β-Arrestin2 in the Brain Cells
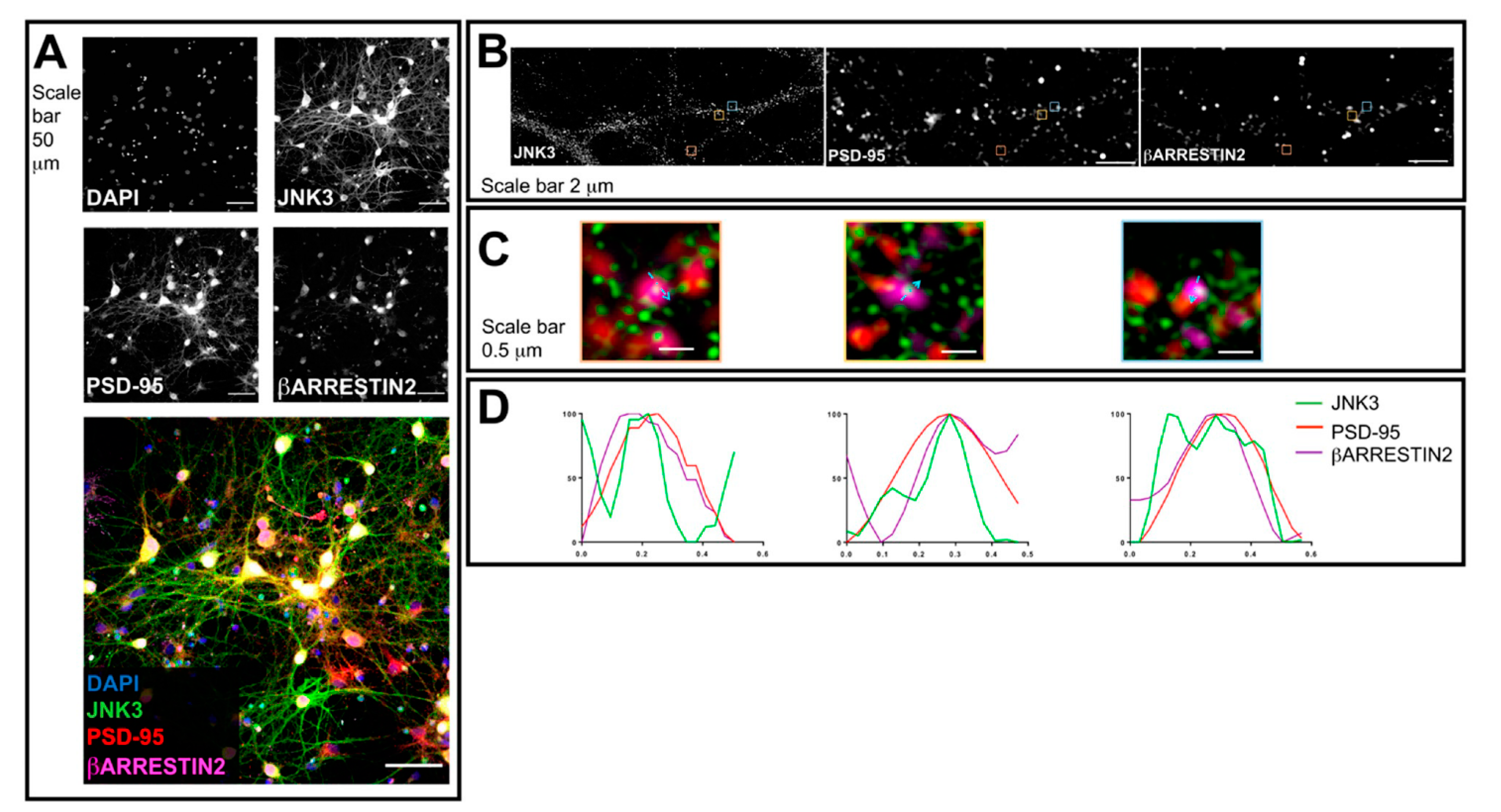
2.3. JNK3 Forms Complexes with PSD95, β-Arrestin2, and JIP1 in Hippocampal Primary Neurons
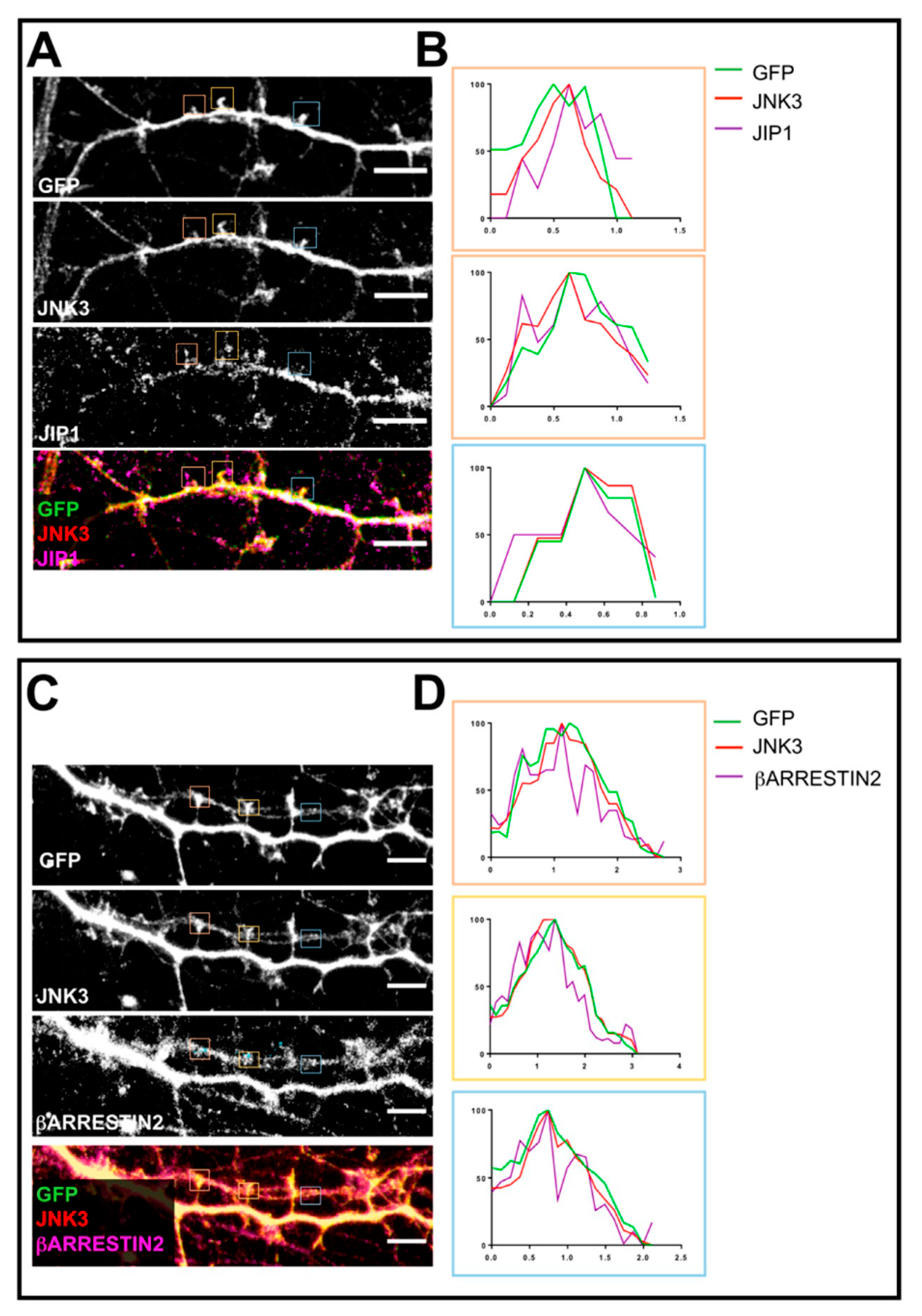
2.4. Localization of JNK3 in Primary Hippocampal Neurons
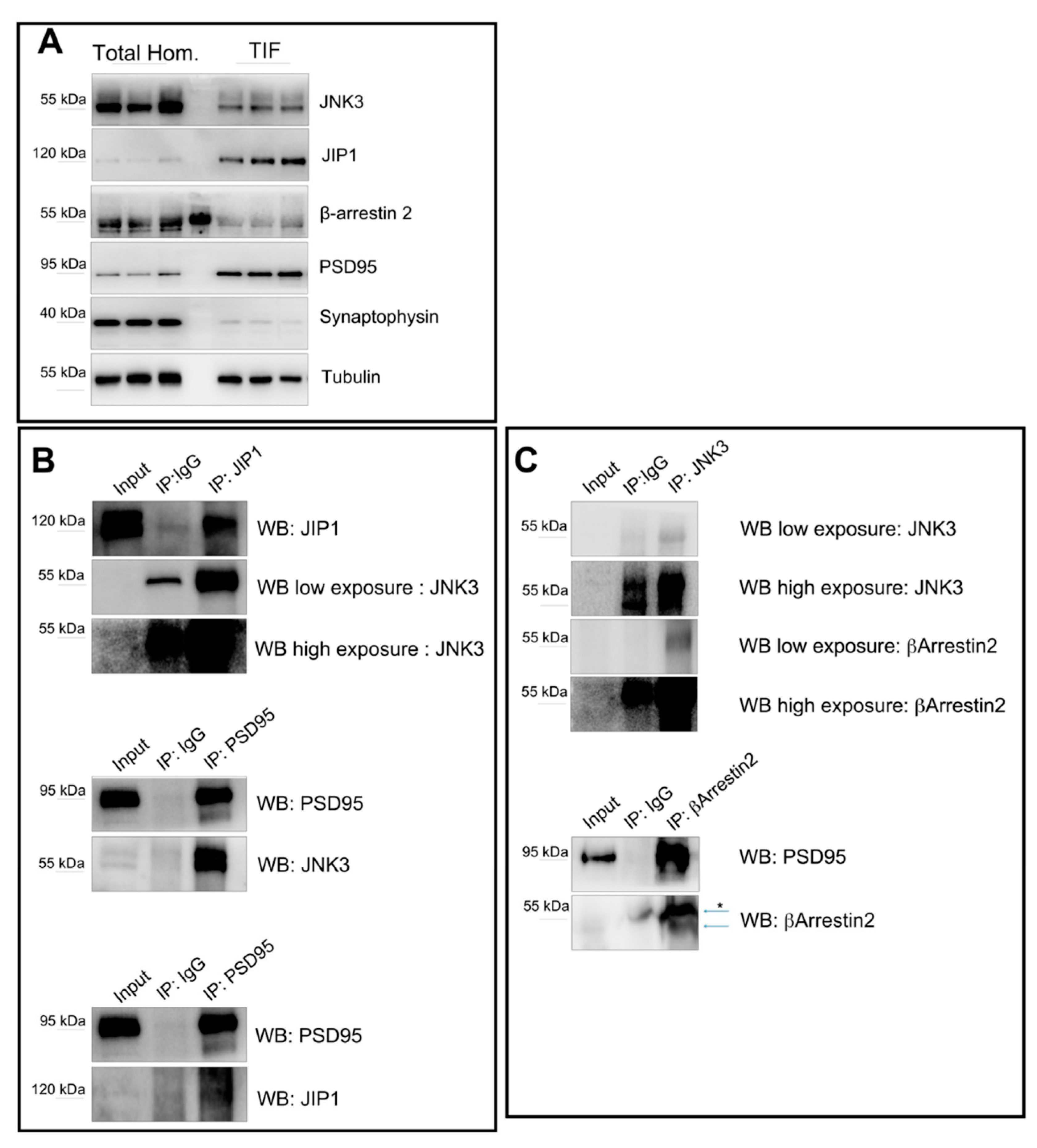
2.5. Interactions of JNK3, JIP1, PSD95, and β-Arrestin2 in Synaptic Fractions
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Hippocampal Primary Neuronal Culture
4.3. Tissue Preparation for Immunoprecipitation Analysis
4.4. Triton-Insoluble Fractionation (TIF)
4.5. Immunoprecipitation
4.6. Western Blotting
4.7. Immunofluorescence
4.8. Confocal Microscopy and Structured Illumination Microscopy (SIM)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.; Narang, H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell. Mol. Life Sci. CMLS 2008, 65, 3525–3544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, C. Regulatory mechanisms of mitogen-activated kinase signaling. Cell. Mol. Life Sci. CMLS 2007, 64, 2771–2789. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, H.J.; Weber, M.J. Mitogen-activated protein kinases: Specific messages from ubiquitous messengers. Mol. Cell. Biol. 1999, 19, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of JNKs: Overcoming the dilemma of protection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef]
- Tenenbaum, M.; Plaisance, V.; Boutry, R.; Pawlowski, V.; Jacovetti, C.; Sanchez-Parra, C.; Ezanno, H.; Bourry, J.; Beeler, N.; Pasquetti, G.; et al. The Map3k12 (Dlk)/JNK3 signaling pathway is required for pancreatic beta-cell proliferation during postnatal development. Cell. Mol. Life Sci. CMLS 2021, 78, 287–298. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Kunde, S.-A.; Rademacher, N.; Tzschach, A.; Wiedersberg, E.; Ullmann, R.; Kalscheuer, V.M.; Shoichet, S.A. Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Hum. Genet. 2013, 132, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K.; Davis, R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Kashef, K.; Lee, C.M.; Xu, H.; Reddy, E.P. Scaffold proteins of MAP-kinase modules. Oncogene 2007, 26, 3185–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickens, M.; Rogers, J.S.; Cavanagh, J.; Raitano, A.; Xia, Z.; Halpern, J.R.; Greenberg, M.E.; Sawyers, C.L.; Davis, R.J. A Cytoplasmic inhibitor of the JNK signal transduction pathway. Science 1997, 277, 693–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmarsh, A.J.; Cavanagh, J.; Tournier, C.; Yasuda, J.; Davis, R.J. A Mammalian scaffold complex that selectively mediates MAP kinase activation. Science 1998, 281, 1671–1674. [Google Scholar] [CrossRef]
- Pellet, J.B.; Haefliger, J.A.; Staple, J.K.; Widmann, C.; Welker, E.; Hirling, H.; Bonny, C.; Nicod, P.; Catsicas, S.; Waeber, G.; et al. Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. Eur. J. Neurosci. 2000, 12, 621–632. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.; Jones, E.V.; Whitmarsh, A.J. The JIP1 scaffold protein regulates axonal development in cortical neurons. Curr. Biol. 2008, 18, 221–226. [Google Scholar] [CrossRef]
- Koushika, S.P. “JIP”ing along the axon: The complex roles of JIPs in axonal transport. BioEssays 2008, 30, 10–14. [Google Scholar] [CrossRef]
- Goldstein, A.Y.; Wang, X.; Schwarz, T.L. Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 2008, 18, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; Holzbaur, E.L.F. JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J. Cell Biol. 2013, 202, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Scheinfeld, M.H.; Roncarati, R.; Vito, P.; Lopez, P.A.; Abdallah, M.; D’Adamio, L. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the alzheimer’s β-amyloid precursor protein (APP). J. Biol. Chem. 2002, 277, 3767–3775. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zhou, L.; Del Villar, K.; Ghanevati, M.; Tashjian, V.; Miller, C.A. JIP1 regulates neuronal apoptosis in response to stress. Mol. Brain Res. 2005, 134, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J.; Kuan, C.-Y.; Kennedy, N.J.; Kelkar, N.; Haydar, T.F.; Mordes, J.P.; Appel, M.; Rossini, A.A.; Jones, S.N.; Flavell, R.A.; et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001, 15, 2421–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, C.; Sherrin, T.; Kennedy, N.J.; Forest, K.H.; Barutcu, S.A.; Robles, M.; Carpenter-Hyland, E.; Alfulaij, N.; Standen, C.L.; Nichols, R.A.; et al. JIP1-mediated JNK activation negatively regulates synaptic plasticity and spatial memory. J. Neurosci. 2018, 38, 3708–3728. [Google Scholar] [CrossRef] [PubMed]
- Harding, T.C.; Xue, L.; Bienemann, A.; Haywood, D.; Dickens, M.; Tolkovsky, A.M.; Uney, J.B. Inhibition of JNK by overexpression of the JNK binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J. Biol. Chem. 2001, 276, 4531–4534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, R.K.; Kendrick, T.S.; Bogoyevitch, M.A. Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 2002, 277, 10987–10997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: Novel blockers of β-cell death. Diabetes 2001, 50, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, J.; Whitmarsh, A.J.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999, 19, 7245–7254. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.H.; Yano, H.; Cho, H.; Meyer, D.; Monks, B.; Margolis, B.; Birnbaum, M.J.; Chao, M.V. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 2002, 35, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Musi, C.A.; Agrò, G.; Santarella, F.; Iervasi, E.; Borsello, T. JNK3 as therapeutic target and biomarker in neurodegenerative and neurodevelopmental brain diseases. Cells 2020, 9, 2190. [Google Scholar] [CrossRef]
- Horiuchi, D.; Barkus, R.V.; Pilling, A.D.; Gassman, A.; Saxton, W.M. APLIP1, a kinesin binding JIP-1/JNK scaffold protein, influences the axonal transport of both vesicles and mitochondria in drosophila. Curr. Biol. 2005, 15, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Margevicius, D.R.; Bastian, C.; Fan, Q.; Davis, R.J.; Pimplikar, S.W. JNK-interacting protein 1 mediates alzheimer’s-like pathological features in AICD-transgenic mice. Neurobiol. Aging 2015, 36, 2370–2379. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Mayor, F. Mechanisms of regulation of the expression and function of g protein-coupled receptor kinases. Cell. Signal. 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Srivastava, A.; Gupta, B.; Gupta, C.; Shukla, A.K. Emerging functional divergence of β-arrestin isoforms in GPCR function. Trends Endocrinol. Metab. TEM 2015, 26, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Green, J.A.; Schmid, C.L.; Bley, E.; Monsma, P.C.; Brown, A.; Bohn, L.M.; Mykytyn, K. Recruitment of β-arrestin into neuronal cilia modulates somatostatin receptor subtype 3 ciliary localization. Mol. Cell. Biol. 2016, 36, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Kook, S.; Gurevich, E.V.; Gurevich, V.V. Arrestin-dependent activation of JNK family kinases. Handb. Exp. Pharmacol. 2014, 219, 259–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Whitmarsh, A.J. The β-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 2008, 283, 15903–15911. [Google Scholar] [CrossRef] [Green Version]
- Breitman, M.; Kook, S.; Gimenez, L.E.; Lizama, B.N.; Palazzo, M.C.; Gurevich, E.V.; Gurevich, V.V. Silent scaffolds: Inhibition of c-Jun N-terminal kinase 3 activity in cell by dominant-negative arrestin-3 mutant. J. Biol. Chem. 2012, 287, 19653–19664. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wu, Y.; Ge, X.; Ma, L.; Pei, G. Subcellular localization of beta-arrestins is determined by their intact N domain and the nuclear export signal at the C terminus. J. Biol. Chem. 2003, 278, 11648–11653. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Bates, M.; Zhuang, X. Super resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, M.; Nozumi, M.; Wu, L.-G.; Zanacchi, F.C.; Katona, I.; Barna, L.; Xu, P.; Zhang, M.; Xue, F.; Boyden, E. New observations in neuroscience using superresolution microscopy. J. Neurosci. 2018, 38, 9459–9467. [Google Scholar] [CrossRef] [PubMed]
- Papa, M.; Bundman, M.C.; Greenberger, V.; Segal, M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 1–11. [Google Scholar] [CrossRef]
- Schikorski, T.; Stevens, C.F. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 5858–5867. [Google Scholar] [CrossRef]
- Gardoni, F.; Schrama, L.H.; Kamal, A.; Gispen, W.H.; Cattabeni, F.; Di Luca, M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Kokotos, A.C.; Harper, C.B.; Marland, J.R.K.; Smillie, K.J.; Cousin, M.A.; Gordon, S.L. Synaptophysin sustains presynaptic performance by preserving vesicular synaptobrevin-II levels. J. Neurochem. 2019, 151, 28–37. [Google Scholar] [CrossRef]
- Borsello, T.; Clarke, P.G.H.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A Peptide inhibitor of C-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef]
- Zhan, X.; Kaoud, T.S.; Dalby, K.N.; Gurevich, V.V. Nonvisual arrestins function as simple scaffolds assembling the MKK4-JNK3α2 signaling complex. Biochemistry 2011, 50, 10520–10529. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Kaoud, T.S.; Kook, S.; Dalby, K.N.; Gurevich, V.V. JNK3 enzyme binding to arrestin-3 differentially affects the recruitment of upstream mitogen-activated protein (MAP) kinase kinases. J. Biol. Chem. 2013, 288, 28535–28547. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Yasukawa, T.; Homma, Y.; Ito, Y.; Niikura, T.; Hiraki, T.; Hirai, S.; Ohno, S.; Kita, Y.; Kawasumi, M.; et al. C-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds alzheimer’s amyloid precursor protein with JNK. J. Neurosci. 2001, 21, 6597–6607. [Google Scholar] [CrossRef] [Green Version]
- Verhey, K.J.; Meyer, D.; Deehan, R.; Blenis, J.; Schnapp, B.J.; Rapoport, T.A.; Margolis, B. Cargo of kinesin identified as jip scaffolding proteins and associated signaling molecules. J. Cell Biol. 2001, 152, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Farías, G.G.; Alfaro, I.E.; Cerpa, W.; Grabowski, C.P.; Godoy, J.A.; Bonansco, C.; Inestrosa, N.C. Wnt-5a/JNK signaling promotes the clustering of PSD-95 in hippocampal neurons. J. Biol. Chem. 2009, 284, 15857–15866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, H.; Vinade, L.; Dosemeci, A. Identification of novel phosphorylation sites on postsynaptic density proteins. Biochem. Biophys. Res. Commun. 2004, 321, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinidad, J.C.; Specht, C.G.; Thalhammer, A.; Schoepfer, R.; Burlingame, A.L. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol. Cell. Proteomics MCP 2006, 5, 914–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Futai, K.; Jo, J.; Hayashi, Y.; Cho, K.; Sheng, M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 2007, 56, 488–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, M.A.; Reddy, P.A. Small molecule JNK (c-Jun N-terminal kinase) inhibitors. J. Med. Chem. 2010, 53, 3005–3012. [Google Scholar] [CrossRef]
- Schnieders, M.J.; Kaoud, T.S.; Yan, C.; Dalby, K.N.; Ren, P. Computational insights for the discovery of non-ATP competitive inhibitors of MAP kinases. Curr. Pharm. Des. 2012, 18, 1173–1185. [Google Scholar] [CrossRef] [Green Version]
- Modi, V.; Dunbrack, R.L. A Structurally-validated multiple sequence alignment of 497 human protein kinase domains. Sci. Rep. 2019, 9, 19790. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.; Gehringer, M.; Laufer, S.A. Inhibitors of C-Jun N-terminal kinases: An update. J. Med. Chem. 2015, 58, 72–95. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Kaoud, T.S.; Yan, C.; Mitra, S.; Tseng, C.-C.; Jose, J.; Taliaferro, J.M.; Tuohetahuntila, M.; Devkota, A.; Sammons, R.; Park, J.; et al. From in silico discovery to intra-cellular activity: Targeting JNK-protein interactions with small molecules. ACS Med. Chem. Lett. 2012, 3, 721–725. [Google Scholar] [CrossRef]
- Sclip, A.; Arnaboldi, A.; Colombo, I.; Veglianese, P.; Colombo, L.; Messa, M.; Mancini, S.; Cimini, S.; Morelli, F.; Antoniou, X.; et al. Soluble Aβ oligomer-induced synaptopathy: C-Jun N-terminal kinase’s role. J. Mol. Cell Biol. 2013, 5, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A. Therapeutic promise of JNK ATP-noncompetitive inhibitors. Trends Mol. Med. 2005, 11, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, A.A.; He, J.; Mou, C.H.; Polak, M.; Zine, A.; Bonny, C.; Balkany, T.J.; Van De Water, T.R. D-JNKI-1 treatment prevents the progression of hearing loss in a model of cochlear implantation trauma. Otol. Neurotol. Off. Publ. Am. Otol. Soc. Am. Neurotol. Soc. Eur. Acad. Otol. Neurotol. 2006, 27, 504–511. [Google Scholar] [CrossRef]
- Eshraghi, A.A.; Aranke, M.; Salvi, R.; Ding, D.; Coleman, J.K.M.; Ocak, E.; Mittal, R.; Meyer, T. Preclinical and clinical otoprotective applications of cell-penetrating peptide D-JNKI-1 (AM-111). Hear. Res. 2018, 368, 86–91. [Google Scholar] [CrossRef]
- Kersting, S.; Behrendt, V.; Kersting, J.; Reinecke, K.; Hilgert, C.; Stricker, I.; Herdegen, T.; Janot, M.S.; Uhl, W.; Chromik, A.M. The impact of JNK inhibitor D-JNKI-1 in a murine model of chronic colitis induced by dextran sulfate sodium. J. Inflamm. Res. 2013, 6, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Milano, G.; Morel, S.; Bonny, C.; Samaja, M.; von Segesser, L.K.; Nicod, P.; Vassalli, G. A peptide inhibitor of C-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1828–H1835. [Google Scholar] [CrossRef] [Green Version]
- Ortolano, F.; Colombo, A.; Zanier, E.R.; Sclip, A.; Longhi, L.; Perego, C.; Stocchetti, N.; Borsello, T.; De Simoni, M.G. C-Jun N-Terminal kinase pathway activation in human and experimental cerebral contusion. J. Neuropathol. Exp. Neurol. 2009, 68, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Sclip, A.; Tozzi, A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.; Salmona, M.; Welker, E.; Borsello, T. C-Jun N-terminal kinase has a key role in alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 2014, 5, e1019. [Google Scholar] [CrossRef]
- Zhao, Y.; Spigolon, G.; Bonny, C.; Culman, J.; Vercelli, A.; Herdegen, T. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell. Neurosci. 2012, 49, 300–310. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Angus, D.W.; Baker, J.A.; Mason, R.; Martin, I.J. The potential influence of CO2, as an agent for euthanasia, on the pharmacokinetics of basic compounds in rodents. Drug Metab. Dispos. Biol. Fate Chem. 2008, 36, 375–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, L.; Carbone, E.T.; Yi, E.M.; Bauer, D.B.; Lindstrom, K.A.; Parker, J.M.; Austin, J.A.; Seo, Y.; Gandhi, A.D.; Wilkerson, J.D. Assessing cervical dislocation as a humane euthanasia method in mice. J. Am. Assoc. Lab. Anim. Sci. JAALAS 2012, 51, 352–356. [Google Scholar] [PubMed]
- Colnaghi, L.; Russo, L.; Natale, C.; Restelli, E.; Cagnotto, A.; Salmona, M.; Chiesa, R.; Fioriti, L. Super resolution microscopy of SUMO proteins in neurons. Front. Cell. Neurosci. 2019, 13, 486. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Natale, C.; Conz, A.; Kelk, J.; Restelli, E.; Chiesa, R.; Salmona, M.; Fioriti, L.; Colnaghi, L. Super-resolution imaging to study co-localization of proteins and synaptic markers in primary neurons. J. Vis. Exp. JoVE 2020, 167. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musi, C.A.; Marchini, G.; Giani, A.; Tomaselli, G.; Priori, E.C.; Colnaghi, L.; Borsello, T. Colocalization and Interaction Study of Neuronal JNK3, JIP1, and β-Arrestin2 Together with PSD95. Int. J. Mol. Sci. 2022, 23, 4113. https://doi.org/10.3390/ijms23084113
Musi CA, Marchini G, Giani A, Tomaselli G, Priori EC, Colnaghi L, Borsello T. Colocalization and Interaction Study of Neuronal JNK3, JIP1, and β-Arrestin2 Together with PSD95. International Journal of Molecular Sciences. 2022; 23(8):4113. https://doi.org/10.3390/ijms23084113
Chicago/Turabian StyleMusi, Clara Alice, Giacomo Marchini, Arianna Giani, Giovanni Tomaselli, Erica Cecilia Priori, Luca Colnaghi, and Tiziana Borsello. 2022. "Colocalization and Interaction Study of Neuronal JNK3, JIP1, and β-Arrestin2 Together with PSD95" International Journal of Molecular Sciences 23, no. 8: 4113. https://doi.org/10.3390/ijms23084113