
Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs


Abstract
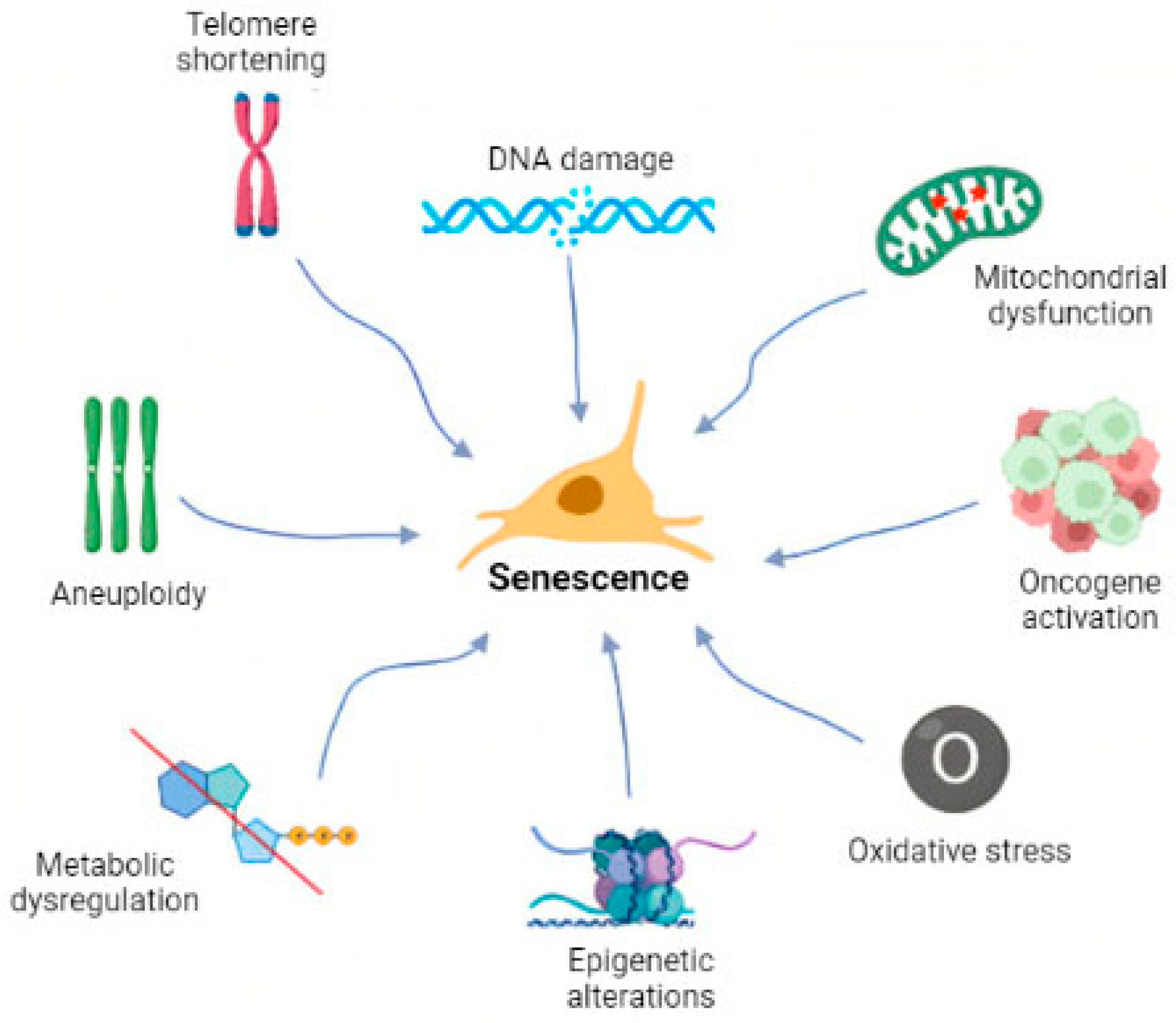
:1. Introduction
2. Senescence Biomarkers and Detection Methods
2.1. Structural Change-Based Markers
Cell Cycle Arrest-Based Markers
2.2. SASP-Associated Markers
2.3. Other Markers
2.4. Probes for Tracing Senescent Cells
3. Cellular Models of Senescence
4. Animal In Vivo Senescence Models
5. Premature Aging Therapy
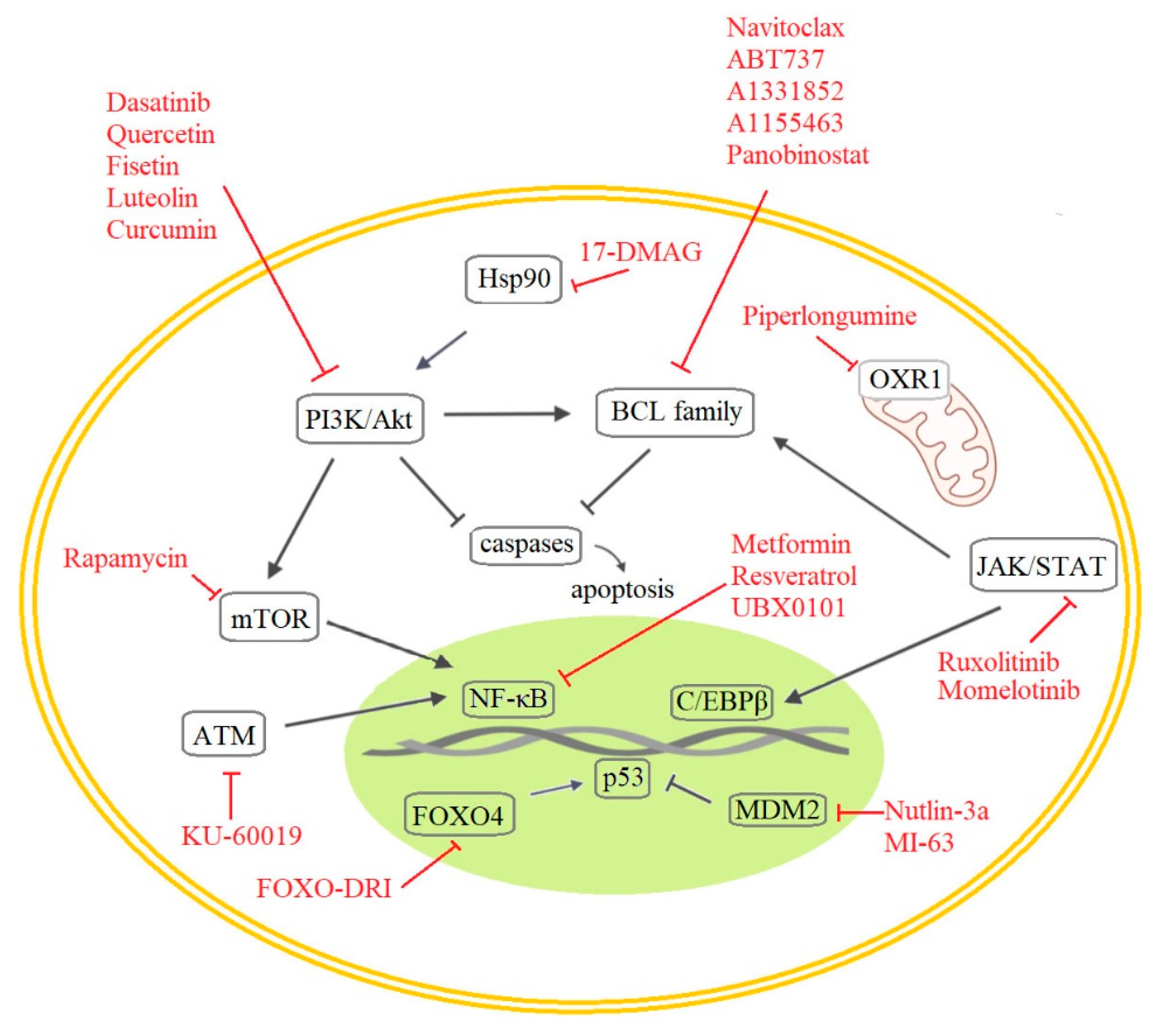
5.1. Senolytic and/or Senotoxic Small Molecules
5.1.1. Anti-Apoptotic Pathways
5.1.2. PI3K and Other Kinases
5.1.3. The p53 and p16 Axis and The DDR Pathway
5.1.4. NF-κB and C/EBPβ Regulation
5.2. Other Approaches to Eliminate Aging Cells
5.3. Senolytics in Anti-Cancer Therapy
5.4. Nanoparticles for Delivery of Active Compounds into Senescent Cells
6. Clinical Trials (CTs)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Search Strategy and Selection Criteria
References
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D. Healing and Hurting: Molecular Mechanisms, Functions, and Pathologies of Cellular Senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The Essence of Senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed Elimination of Senescent Cells by Inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Hubackova, S.; Davidova, E.; Rohlenova, K.; Stursa, J.; Werner, L.; Andera, L.; Dong, L.F.; Terp, M.G.; Hodny, Z.; Ditzel, H.J.; et al. Selective Elimination of Senescent Cells by Mitochondrial Targeting Is Regulated by ANT2. Cell Death Differ. 2019, 26, 276–290. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of Senescent Cells by β-Galactosidase-Targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative Stress, Aging, and Diseases. Clin. Interv. Aging 2018, 13, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and Senescence: A New Insight in Selected Human Diseases. J. Cell. Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.D.; Lapi, E.; Sullivan, A.; Jia, W.; He, Y.W.; Ratnayaka, I.; Zhong, S.; Goldin, R.D.; Goemans, C.G.; et al. Autophagic Activity Dictates the Cellular Response to Oncogenic RAS. Proc. Natl. Acad. Sci. USA 2012, 109, 13325–13330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565.e3. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The Activities of Senescence in Cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [Green Version]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy Maintains Stemness by Preventing Senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and Aging: The Critical Roles of P53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor P21cip1/Waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [Green Version]
- Benson, E.K.; Mungamuri, S.K.; Attie, O.; Kracikova, M.; Sachidanandam, R.; Manfredi, J.J.; Aaronson, S.A. P53-Dependent Gene Repression through P21 Is Mediated by Recruitment of E2F4 Repression Complexes. Oncogene 2014, 33, 3959–3969. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a Signature of in vivo Senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of P16INK4a and P21CIP1 Pathways to Induction of Premature Senescence of Human Endothelial Cells: Permissive Role of P53. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H1575–H1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentino, F.P.; Symonds, C.E.; MacAluso, M.; Giordano, A. Senescence and P130/Rbl2: A New Beginning to the End. Cell Res. 2009, 19, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular Senescence and Tumor Suppressor Gene P16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Helmbold, H.; Kömm, K.; Deppert, W.; Bohn, W. Rb2/P130 Is the Dominating Pocket Protein in the P53–P21 DNA Damage Response Pathway Leading to Senescence. Oncogene 2009, 28, 3456–3467. [Google Scholar] [CrossRef] [Green Version]
- Indovina, P.; Marcelli, E.; Casini, N.; Rizzo, V.; Giordano, A. Emerging Roles of RB Family: New Defense Mechanisms against Tumor Progression. J. Cell. Physiol. 2013, 228, 525–535. [Google Scholar] [CrossRef]
- Helmbold, H.; Deppert, W.; Bohn, W. Regulation of Cellular Senescence by Rb2/P130. Oncogene 2006, 25, 5257–5262. [Google Scholar] [CrossRef] [Green Version]
- Rayman, J.B.; Takahashi, Y.; Indjeian, V.B.; Dannenberg, J.H.; Catchpole, S.; Watson, R.J.; Riele, H.; Dynlacht, B.D. E2F Mediates Cell Cycle-Dependent Transcriptional Repression in vivo by Recruitment of an HDAC1/MSin3B Corepressor Complex. Genes Dev. 2002, 16, 933–947. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, B.B.; Blasco, M.A. Assessing Cell and Organ Senescence Biomarkers. Circ. Res. 2012, 111, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noren Hooten, N.; Evans, M.K. Techniques to Induce and Quantify Cellular Senescence. J. Vis. Exp. 2017, 2017, 55533. [Google Scholar] [CrossRef] [PubMed]
- Adewoye, A.B.; Tampakis, D.; Follenzi, A.; Stolzing, A. Multiparameter Flow Cytometric Detection and Quantification of Senescent Cells in vitro. Biogerontology 2020, 21, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Flenkenthaler, F.; Stöckl, J.B.; Dietrich, K.G.; Köhn, F.M.; Schwarzer, J.U.; Kunz, L.; Luckner, M.; Wanner, G.; Arnold, G.J.; et al. Insights into Replicative Senescence of Human Testicular Peritubular Cells. Sci. Rep. 2019, 9, 15052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Βgal) Activity, a Biomarker of Senescent Cells in Culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Itahana, K.; Itahana, Y.; Dimri, G.P. Colorimetric Detection of Senescence-Associated β Galactosidase. Methods Mol. Biol. 2013, 965, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is Senescence-Associated β-Galactosidase a Marker of Neuronal Senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef] [Green Version]
- de Mera-Rodríguez, J.A.; Álvarez-Hernán, G.; Gañán, Y.; Martín-Partido, G.; Rodríguez-León, J.; Francisco-Morcillo, J. Is Senescence-Associated β-Galactosidase a Reliable in vivo Marker of Cellular Senescence during Embryonic Development? Front. Cell Dev. Biol. 2021, 9, 36. [Google Scholar] [CrossRef]
- Hildebrand, D.G.; Lehle, S.; Borst, A.; Haferkamp, S.; Essmann, F.; Schulze-Osthoff, K. α-Fucosidase as a Novel Convenient Biomarker for Cellular Senescence. Cell Cycle 2013, 12, 1922–1927. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Brunk, U.T. Lipofuscin: Mechanisms of Formation and Increase with Age. APMIS 1998, 106, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [CrossRef] [PubMed]
- Salmonowicz, H.; Passos, J.F. Detecting Senescence: A New Method for an Old Pigment. Aging Cell 2017, 16, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. ΓH2AX as a Molecular Marker of Aging and Disease. Epigenetics 2010, 5, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jager, M.; Dronkert, M.L.G.; Modesti, M.; Beerens, C.E.M.T.; Kanaar, R.; Van Gent, D.C. DNA-Binding and Strand-Annealing Activities of Human Mre11: Implications for Its Roles in DNA Double-Strand Break Repair Pathways. Nucleic Acids Res. 2001, 29, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Matsuoka, S.; Carpenter, P.B.; Elledge, S.J. 53BP1, a Mediator of the DNA Damage Checkpoint. Science 2002, 298, 1435–1438. [Google Scholar] [CrossRef]
- Stucki, M.; Jackson, S.P. ΓH2AX and MDC1: Anchoring the DNA-Damage-Response Machinery to Broken Chromosomes. DNA Repair 2006, 5, 534–543. [Google Scholar] [CrossRef]
- Takai, H.; Smogorzewska, A.; De Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21CIP1, but Not P16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Olovnikov, A.M. Telomeres, Telomerase, and Aging: Origin of the Theory. Exp. Gerontol. 1996, 31, 443–448. [Google Scholar] [CrossRef]
- Parrinello, S.; Samper, E.; Krtolica, A.; Goldstein, J.; Melov, S.; Campisi, J. Oxygen Sensitivity Severely Limits the Replicative Lifespan of Murine Fibroblasts. Nat. Cell Biol. 2003, 5, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria Are Required for Pro-Ageing Features of the Senescent Phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Pole, A.; Dimri, M.; Dimri, G.P. Oxidative Stress, Cellular Senescence and Ageing. AIMS Mol. Sci. 2016, 3, 300–324. [Google Scholar] [CrossRef]
- Agrawal, K.; Das, V.; Táborská, N.; Gurský, J.; Džubák, P.; Hajdúch, M. Differential Regulation of Methylation-Regulating Enzymes by Senescent Stromal Cells Drives Colorectal Cancer Cell Response to DNA-Demethylating Epi-Drugs. Stem Cells Int. 2018, 2018, 6013728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, K.M.; Zhang, R. Detection of Senescence-Associated Heterochromatin Foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular Dissection of Formation of Senescence-Associated Heterochromatin Foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Muñoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppé, J.P.; Campeau, E.; Beauséjour, C.M.; Kim, S.H.; et al. DNA-SCARS: Distinct Nuclear Structures That Sustain Damage-Induced Senescence Growth Arrest and Inflammatory Cytokine Secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Gratzner, H.G. Monoclonal Antibody to 5-Bromo- and 5-Iododeoxyuridine: A New Reagent for Detection of DNA Replication. Science 1982, 218, 474–475. [Google Scholar] [CrossRef]
- Buck, S.B.; Bradford, J.; Gee, K.R.; Agnew, B.J.; Clarke, S.T.; Salic, A. Detection of S-Phase Cell Cycle Progression Using 5-Ethynyl-2′-Deoxyuridine Incorporation with Click Chemistry, an Alternative to Using 5-Bromo-2′-Deoxyuridine Antibodies. Biotechniques 2008, 44, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Gerdes, J. The Ki-67 Protein: From the Known and the Unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Zhang, H. Molecular Signaling and Genetic Pathways of Senescence: Its Role in Tumorigenesis and Aging. J. Cell. Physiol. 2007, 210, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, J.; Yan, B.; Chen, X. DEC1, a Basic Helix-Loop-Helix Transcription Factor and a Novel Target Gene of the P53 Family, Mediates P53-Dependent Premature Senescence*|Elsevier Enhanced Reader. J. Biol. Chem. 2008, 283, 2896–2905. Available online: https://reader.elsevier.com/reader/sd/pii/S0021925820555385?token=088C94F7A5AE15857D34F420A29A72A3BE5345F5D1D288FFC595813C13C107D9B6E1F7759CFB209D116494F9DD7EAC6D&originRegion=eu-west-1&originCreation=20210412194802 (accessed on 12 April 2021). [CrossRef] [PubMed] [Green Version]
- Ruiz, L.; Traskine, M.; Ferrer, I.; Castro, E.; Leal, J.F.M.; Kaufman, M.; Carnero, A. Characterization of the P53 Response to Oncogene-Induced Senescence. PLoS ONE 2008, 3, e3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (Sasp) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Mrazkova, B.; Dzijak, R.; Imrichova, T.; Kyjacova, L.; Barath, P.; Dzubak, P.; Holub, D.; Hajduch, M.; Nahacka, Z.; Andera, L.; et al. Induction, Regulation and Roles of Neural Adhesion Molecule L1CAM in Cellular Senescence. Aging 2018, 10, 434–462. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of Novel Markers of Senescence and Their Prognostic Potential in Cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [Green Version]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Heo, C.H.; Sen, D.; Byun, H.O.; Kwak, I.H.; Yoon, G.; Kim, H.M. Ratiometric Two-Photon Fluorescent Probe for Quantitative Detection of β-Galactosidase Activity in Senescent Cells. Anal. Chem. 2014, 86, 10001–10005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, C.; Dutta, C.; Fang, M.; Zhang, S.; Tiwari, A.; Werner, T.; Luo, F.T.; Liu, H. A Novel Near-Infrared Fluorescent Probe for Sensitive Detection of β-Galactosidase in Living Cells. Anal. Chim. Acta 2017, 968, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano-Torres, B.; Galiana, I.; Rovira, M.; Garrido, E.; Chaib, S.; Bernardos, A.; Muñoz-Espín, D.; Serrano, M.; Martínez-Máñez, R.; Sancenón, F. An OFF-ON Two-Photon Fluorescent Probe for Tracking Cell Senescence in vivo. J. Am. Chem. Soc. 2017, 139, 8808–8811. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Ma, X.; Cui, C.; Deenik, P.R.; Henderson, P.K.P.; Sigler, A.L.; Cui, L. Real-Time Imaging of Senescence in Tumors with DNA Damage. Sci. Rep. 2019, 9, 2102. [Google Scholar] [CrossRef] [Green Version]
- Becker, T.; Haferkamp, S. Molecular Mechanisms of Cellular Senescence. In Senescence and Senescence-Related Disorders; InTech: Vienna, Austria, 2013. [Google Scholar]
- Eriksson, D.; Stigbrand, T. Radiation-Induced Cell Death Mechanisms. Tumor Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, S.; Liu, Y.; Xu, H.; Liang, J.; Zhu, J.; Zhang, G.; Su, W.; Dong, W.; et al. Upregulation of EID3 Sensitizes Breast Cancer Cells to Ionizing Radiation-Induced Cellular Senescence. Biomed. Pharmacother. 2018, 107, 606–614. [Google Scholar] [CrossRef]
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat. Res. 2016, 186, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Dolan, D.W.P.; Zupanic, A.; Nelson, G.; Hall, P.; Miwa, S.; Kirkwood, T.B.L.; Shanley, D.P. Integrated Stochastic Model of DNA Damage Repair by Non-Homologous End Joining and P53/P21- Mediated Early Senescence Signalling. PLoS Comput. Biol. 2015, 11, e1004246. [Google Scholar] [CrossRef]
- Day, R.M.; Snow, A.L.; Panganiban, R.A.M. Radiation-Induced Accelerated Senescence: A Fate Worse than Death? Cell Cycle 2014, 13, 2011–2012. [Google Scholar] [CrossRef] [Green Version]
- Gladyshev, V.N. The Free Radical Theory of Aging Is Dead. Long Live the Damage Theory! Antioxid. Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA Damage and Senescence of Human Diploid Fibroblast Cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular Analysis of H2O2-Induced Senescent-like Growth Arrest in Normal Human Fibroblasts: P53 and Rb Control G1 Arrest but Not Cell Replication. Biochem. J. 1998, 332, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-H.; Ozanne, S.E.; Hales, C.N. Methods of Cellular Senescence Induction Using Oxidative Stress. In Biological Aging; Humana Press: Totowa, NJ, USA, 2007; pp. 179–189. [Google Scholar]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and P16(INK4a). Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Wajapeyee, N. Induction of Cellular Senescence by Oncogenic RAS. Methods Mol. Biol. 2013, 1048, 127–133. [Google Scholar] [CrossRef]
- Zhang, F.; Zakaria, S.M.; Högqvist Tabor, V.; Singh, M.; Tronnersjö, S.; Goodwin, J.; Selivanova, G.; Bartek, J.; Castell, A.; Larsson, L.G. MYC and RAS Are Unable to Cooperate in Overcoming Cellular Senescence and Apoptosis in Normal Human Fibroblasts. Cell Cycle 2018, 17, 2697–2715. [Google Scholar] [CrossRef]
- The NIA Aging Cell Repository: Facilitating Research with Aging Cells; National Institute on Aging: Bethesda, MD, USA. Available online: https://www.nia.nih.gov/research/blog/2018/05/nia-aging-cell-repository-facilitating-research-aging-cells (accessed on 19 April 2021).
- Kudlow, B.A.; Kennedy, B.K.; Monnat, R.J. Werner and Hutchinson-Gilford Progeria Syndromes: Mechanistic Basis of Human Progeroid Diseases. Nat. Rev. Mol. Cell Biol. 2007, 8, 394–404. [Google Scholar] [CrossRef]
- Harkema, L.; Youssef, S.A.; De Bruin, A. Pathology of Mouse Models of Accelerated Aging. Vet. Pathol. 2016, 53, 366–389. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Melos, K.I.; Angelini, L.; Burd, C.E.; Robbins, P.D.; Niedernhofer, L.J. Mouse Models of Accelerated Cellular Senescence. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1896, pp. 203–230. [Google Scholar]
- Sorrentino, J.A.; Krishnamurthy, J.; Tilley, S.; Alb, J.G.; Burd, C.E.; Sharpless, N.E. P16INK4a Reporter Mice Reveal Age-Promoting Effects of Environmental Toxicants. J. Clin. Investig. 2014, 124, 169–173. [Google Scholar] [CrossRef]
- Le, O.N.L.; Rodier, F.; Fontaine, F.; Coppe, J.P.; Campisi, J.; DeGregori, J.; Laverdière, C.; Kokta, V.; Haddad, E.; Beauséjour, C.M. Ionizing Radiation-Induced Long-Term Expression of Senescence Markers in Mice Is Independent of P53 and Immune Status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, M.-A.; Jung, U.; Eom, S.; Kim, S.-H.; Park, H.-R.; Jo, S.-K. Chronic Induction of Senescence Marker in Gamma-Irradiation Mice. In Proceedings of the Transactions of the Korean Nuclear Society Autumn Meeting, Gyeongju, Korea, 27–28 October 2011. [Google Scholar]
- Seol, M.A.; Jung, U.; Eom, H.S.; Kim, S.H.; Park, H.R.; Jo, S.K. Prolonged Expression of Senescence Markers in Mice Exposed to Gamma-Irradiation. J. Vet. Sci. 2012, 13, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Hei, T.K. Aging and Age-Related Health Effects of Ionizing Radiation. Radiat. Med. Prot. 2020, 1, 15–23. [Google Scholar] [CrossRef]
- Kudlova, N.; Slavik, H.; Duskova, P.; Furst, T.; Srovnal, J.; Bartek, J.; Mistrik, M.; Hajduch, M. An Efficient, Non-Invasive Approach for in-vivo Sampling of Hair Follicles: Design and Applications in Monitoring DNA Damage and Aging. Aging 2021, 13, 25004–25024. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Du, M.; Dolence, E.K.; Fang, C.X.; Mayer, G.E.; Ceylan-Isik, A.F.; LaCour, K.H.; Yang, X.; Wilbert, C.J.; Sreejayan, N.; et al. Aging Induces Cardiac Diastolic Dysfunction, Oxidative Stress, Accumulation of Advanced Glycation Endproducts and Protein Modification. Aging Cell 2005, 4, 57–64. [Google Scholar] [CrossRef]
- Yoo, D.Y.; Kim, W.; Lee, C.H.; Shin, B.N.; Nam, S.M.; Choi, J.H.; Won, M.H.; Yoon, Y.S.; Hwang, I.K. Melatonin Improves D-Galactose-Induced Aging Effects on Behavior, Neurogenesis, and Lipid Peroxidation in the Mouse Dentate Gyrus via Increasing PCREB Expression. J. Pineal Res. 2012, 52, 21–28. [Google Scholar] [CrossRef]
- Salehpour, F.; Ahmadian, N.; Rasta, S.H.; Farhoudi, M.; Karimi, P.; Sadigh-Eteghad, S. Transcranial Low-Level Laser Therapy Improves Brain Mitochondrial Function and Cognitive Impairment in D-Galactose–Induced Aging Mice. Neurobiol. Aging 2017, 58, 140–150. [Google Scholar] [CrossRef]
- Remigante, A.; Spinelli, S.; Trichilo, V.; Loddo, S.; Sarikas, A.; Pusch, M.; Dossena, S.; Marino, A.; Morabito, R. D-Galactose Induced Early Aging in Human Erythrocytes: Role of Band 3 Protein. J. Cell. Physiol. 2022, 237, 1586–1596. [Google Scholar] [CrossRef]
- Sun, K.; Yang, P.; Zhao, R.; Bai, Y.; Guo, Z. Matrine Attenuates D-Galactose-Induced Aging-Related Behavior in Mice via Inhibition of Cellular Senescence and Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 7108604. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, Y.; Guo, Y.; Xu, L.; Wang, H. Phlorizin Exerts Potent Effects against Aging Induced by D-Galactose in Mice and PC12 Cells. Food Funct. 2021, 12, 2148–2160. [Google Scholar] [CrossRef]
- Li, J.-H.; Wei, T.-T.; Guo, L.; Cao, J.-H.; Feng, Y.-K.; Guo, S.-N.; Liu, G.-H.; Ding, Y.; Chai, Y.-R. Curcumin Protects Thymus against D-Galactose-Induced Senescence in Mice. Naunyn. Schmiedebergs. Arch. Pharmacol. 2021, 394, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Liang, C.J.; Weng, Y.W.; Chen, Y.H.; Hsu, H.Y.; Chien, H.F.; Tsai, J.S.; Tseng, Y.C.; Li, C.Y.; Chen, Y.L. Ganoderma Lucidum Polysaccharides Prevent Platelet-Derived Growth Factor-Stimulated Smooth Muscle Cell Proliferation in vitro and Neointimal Hyperplasia in the Endothelial-Denuded Artery in vivo. J. Cell. Physiol. 2012, 227, 3063–3071. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.S.; Kim, E.; Kim, Y.; Kim, Y. Curcumin and Hesperetin Attenuate D-Galactose-Induced Brain Senescence in vitro and in vivo. Nutr. Res. Pract. 2020, 14, 438. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Chen, W.; Nong, Z.; Huang, J.; Chen, C. Protective Effect of Hyperbaric Oxygen on Cognitive Impairment Induced by D-Galactose in Mice. Neurochem. Res. 2016, 41, 3032–3041. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-Dependent Oncogene-Induced Senescence in vivo and Its Evasion during Mammary Tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef]
- Baek, K.H.; Ryeom, S. Detection of Oncogene-Induced Senescence in vivo. Methods Mol. Biol. 2017, 1534, 185–198. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific Inhibition of Cyclin-Dependent Kinase 4/6 by PD 0332991 and Associated Antitumor Activity in Human Tumor Xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical Characterization of the CDK4/6 Inhibitor LY2835219: In-Vivo Cell Cycle-Dependent/Independent Anti-Tumor Activities Alone/in Combination with Gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Loo, A.; Chopra, R.; Caponigro, G.; Huang, A.; Vora, S.; Parasuraman, S.; Howard, S.; Keen, N.; Sellers, W.; et al. Abstract PR02: LEE011: An Orally Bioavailable, Selective Small Molecule Inhibitor of CDK4/6– Reactivating Rb in Cancer. Mol. Cancer Ther. 2013, 12, PR02. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Decristo, M.J.; Watt, A.C.; Brinjones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollard, J.; Miguela, V.; Ruiz De Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sánchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a Selective CDK4/6 Inhibitor, Restricts Tumour Growth in Preclinical Models of Hepatocellular Carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef] [Green Version]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 Inhibition Induces Cell-Cycle Arrest and Senescence in Neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-Induced Autophagy and Senescence in Gastric Cancer Cells. Exp. Cell Res. 2017, 360, 390–396. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liao, W.; Klein, M.E.; Robine, N.; Geiger, H.; Crago, A.M.; Dickson, M.A.; Tap, W.D.; Singer, S.; Koff, A. ATRX Is a Regulator of Therapy Induced Senescence in Human Cells. Nat. Commun. 2017, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, C.; Miao, Z.; Wu, Y.; Guo, Z.; Li, J.; Yao, J.; Xing, C.; Sheng, C.; Zhang, W. Double-Edged Swords as Cancer Therapeutics: Novel, Orally Active, Small Molecules Simultaneously Inhibit P53-MDM2 Interaction and the NF-ΚB Pathway. J. Med. Chem. 2014, 57, 567–577. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liu, D.D.; Dickson, M.A.; Klein, M.E.; O’Connor, R.; Wilder, F.O.; Socci, N.D.; Tap, W.D.; Schwartz, G.K.; Singer, S.; et al. MDM2 Turnover and Expression of ATRX Determine the Choice between Quiescence and Senescence in Response to CDK4 Inhibition. Oncotarget 2015, 6, 8226–8243. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Varela-Eirin, M.; Brandenburg, S.M.; Hernandez-Segura, A.; van Vliet, T.; Jongbloed, E.M.; Wilting, S.M.; Ohtani, N.; Jager, A.; Demaria, M. Pharmacological CDK4/6 Inhibition Reveals a P53-Dependent Senescent State with Restricted Toxicity. EMBO J. 2022, 41, e108946. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Ma, B.B.Y.; Hui, C.W.C.; Lo, K.W.; Hui, E.P.; Chan, A.T.C. Preclinical Evaluation of Ribociclib and Its Synergistic Effect in Combination with Alpelisib in Non-Keratinizing Nasopharyngeal Carcinoma. Sci. Rep. 2018, 8, 8010. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic Drugs: From Discovery to Translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Targeting Normal and Cancer Senescent Cells as a Strategy of Senotherapy. Ageing Res. Rev. 2019, 55, 100941. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy HHS Public Access. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Send in the Senolytics. Nat. Biotechnol. 2020, 38, 1371–1377. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and Apoptosis: Dueling or Complementary Cell Fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor Suppressor and Aging Biomarker P16(INK4a) Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [Green Version]
- Burton, D.G.A.; Stolzing, A. Cellular Senescence: Immunosurveillance and Future Immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, G.; Dellambra, E.; Paterna, P.; Golisano, O.; Traverso, C.E.; Rama, P.; Lacal, P.; De Luca, M. Telomerase Activity Is Sufficient to Bypass Replicative Senescence in Human Limbal and Conjunctival but Not Corneal Keratinocytes. Eur. J. Cell Biol. 2004, 83, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic P53-Independent P21 Expression Causes Genomic Instability by Deregulating Replication Licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ät-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating Senescent and Centenarian Human Cells by Reprogramming through the Pluripotent State. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of HTERT Gene Expression Promotes Escape from Oncogene-Induced Cellular Senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-Associated Reprogramming Promotes Cancer Stemness. Nature 2017, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and Senostatics as Adjuvant Tumour Therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA Damage and Senescence in Osteoprogenitors Expressing Osx1 May Cause Their Decrease with Age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (Venetoclax) and BCL-2 Inhibitors in Clinical Development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New Agents That Target Senescent Cells: The Flavone, Fisetin, and the BCL-XL Inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wild, C.; Ding, Y.; Ye, N.; Chen, H.; Wold, E.A.; Zhou, J. BH4 Domain of Bcl-2 as a Novel Target for Cancer Therapy. Drug Discov. Today 2016, 21, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Y.; Zhang, X.; Gao, Z.; Zhang, S.; Shi, P.; Zhang, X.; Song, L.; Hendrickson, H.; Zhou, D.; et al. Senolytic Activity of Piperlongumine Analogues: Synthesis and Biological Evaluation. Bioorg. Med. Chem. 2018, 26, 3925–3938. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of Piperlongumine as a Potential Novel Lead for the Development of Senolytic Agents. Aging 2016, 8, 2915. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, S.; Liu, X.; Wang, Y.; Chang, J.; Zhang, X.; Mackintosh, S.G.; Tackett, A.J.; He, Y.; Lv, D.; et al. Oxidation Resistance 1 Is a Novel Senolytic Target. Aging Cell 2018, 17, 12780. [Google Scholar] [CrossRef]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin Is a Senotherapeutic That Extends Health and Lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tia, N.; Singh, A.K.; Pandey, P.; Azad, C.S.; Chaudhary, P.; Gambhir, I.S. Role of Forkhead Box O (FOXO) Transcription Factor in Aging and Diseases. Gene 2018, 648, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 Inhibitors as a Novel Class of Senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical Screening Identifies ATM as a Target for Alleviating Senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging Role of NF-ΚB Signaling in the Induction of Senescence-Associated Secretory Phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. MTOR Regulates MAPKAPK2 Translation to Control the Senescence-Associated Secretory Phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Alimbetov, D.; Davis, T.; Brook, A.J.C.; Cox, L.S.; Faragher, R.G.A.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the Senescence-Associated Secretory Phenotype (SASP) in Human Fibroblasts Using Small Molecule Inhibitors of P38 MAP Kinase and MK2. Biogerontology 2016, 17, 305. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-Molecule MDM2 Antagonists Attenuate the Senescence-Associated Secretory Phenotype. Sci. Rep. 2018, 8, 2410. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local Clearance of Senescent Cells Attenuates the Development of Post-Traumatic Osteoarthritis and Creates a pro-Regenerative Environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin Inhibits the Senescence-Associated Secretory Phenotype by Interfering with IKK/NF-ΚB Activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Pitozzi, V.; Mocali, A.; Laurenzana, A.; Giannoni, E.; Cifola, I.; Battaglia, C.; Chiarugi, P.; Dolara, P.; Giovannelli, L. Chronic Resveratrol Treatment Ameliorates Cell Adhesion and Mitigates the Inflammatory Phenotype in Senescent Human Fibroblasts. J. Gerontol. A. Biol. Sci. Med. Sci. 2013, 68, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK Inhibition Alleviates the Cellular Senescence-Associated Secretory Phenotype and Frailty in Old Age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [Green Version]
- Van Rhee, F.; Wong, R.S.; Munshi, N.; Rossi, J.F.; Ke, X.Y.; Fosså, A.; Simpson, D.; Capra, M.; Liu, T.; Hsieh, R.K.; et al. Siltuximab for Multicentric Castleman’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2014, 15, 966–974. [Google Scholar] [CrossRef]
- Raffaele, M.; Kovacovicova, K.; Biagini, T.; Lo Re, O.; Frohlich, J.; Giallongo, S.; Nhan, J.D.; Giannone, A.G.; Cabibi, D.; Ivanov, M.; et al. Nociceptin/Orphanin FQ Opioid Receptor (NOP) Selective Ligand MCOPPB Links Anxiolytic and Senolytic Effects. GeroScience 2022, 44, 463–483. [Google Scholar] [CrossRef]
- Cochemé, H.M.; Murphy, M.P. Can Antioxidants Be Effective Therapeutics? Curr. Opin. Investig. Drugs 2010, 11, 426–431. Available online: https://pubmed.ncbi.nlm.nih.gov/20336590/ (accessed on 20 March 2022).
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant Therapy: Current Status and Future Prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in Randomized Trials of Antioxidant Supplements for Primary and Secondary Prevention: Systematic Review and Meta-Analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef]
- Dallner, G.; Sindelar, P.J. Regulation of Ubiquinone Metabolism. Free Radic. Biol. Med. 2000, 29, 285–294. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Ávila, M.; Fernández Vega, A.; De La Mata, M.; Delgado Pavón, A.; De Miguel, M.; Pérez Calero, C.; Villanueva Paz, M.; Cotán, D.; et al. Coenzyme Q10 Therapy. Mol. Syndromol. 2014, 5, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, M.A.; Hoffmann, P.R. The Human Selenoproteome: Recent Insights into Functions and Regulation. Cell. Mol. Life Sci. 2009, 66, 2457–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinceti, M.; Bonvicini, F.; Bergomi, M.; Malagoli, C. Possible Involvement of Overexposure to Environmental Selenium in the Etiology of Amyotrophic Lateral Sclerosis: A Short Review. Ann. Dell’istituto Super. Sanità 2010, 46, 279–283. [Google Scholar] [CrossRef]
- Sanmartin, C.; Plano, D.; Font, M.; Palop, J.A. Selenium and Clinical Trials: New Therapeutic Evidence for Multiple Diseases. Curr. Med. Chem. 2011, 18, 4635–4650. [Google Scholar] [CrossRef] [PubMed]
- Artero, A.; Artero, A.; Tarín, J.J.; Cano, A. The Impact of Moderate Wine Consumption on Health. Maturitas 2015, 80, 3–13. [Google Scholar] [CrossRef]
- Bouzid, M.A.; Filaire, E.; McCall, A.; Fabre, C. Radical Oxygen Species, Exercise and Aging: An Update. Sports Med. 2015, 45, 1245–1261. [Google Scholar] [CrossRef]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen Consumption and Usage during Physical Exercise: The Balance between Oxidative Stress and ROS-Dependent Adaptive Signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [Green Version]
- Battram, A.M.; Bachiller, M.; Martín-Antonio, B. Senescence in the Development and Response to Cancer with Immunotherapy: A Double-Edged Sword. Int. J. Mol. Sci. 2020, 21, 4346. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, A.; Burton, D.G.A.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D Ligands Mediate Immunosurveillance of Senescent Cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kale, A.; Sharma, A.; Stolzing, A.; Stolzing, A.; Desprez, P.Y.; Desprez, P.Y.; Campisi, J.; Campisi, J. Role of Immune Cells in the Removal of Deleterious Senescent Cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired Immune Surveillance Accelerates Accumulation of Senescent Cells and Aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutsatsou, P.; Ochs, J.; Schmitt, R.H.; Hewitt, C.J.; Hanga, M.P. Automation in Cell and Gene Therapy Manufacturing: From Past to Future. Biotechnol. Lett. 2019, 41, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. P53-Dependent Chemokine Production by Senescent Tumor Cells Supports NKG2D-Dependent Tumor Elimination by Natural Killer Cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Passos, J.F. Targeting the SASP to Combat Ageing: Mitochondria as Possible Intracellular Allies? Bioessays 2017, 39, 1600235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Leite de Oliveira, R.; Wang, C.; Fernandes Neto, J.M.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep. 2017, 21, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A Delineation of Cancer Cell Senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of Therapy-Induced Senescent Tumor Cells by the Senolytic ABT-263 via Interference with BCL-X L -BAX Interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Schiff, D.S.; Lin, Y.; Neboori, H.J.R.; Goyal, S.; Feng, Z.; Haffty, B.G. Ionizing Radiation Sensitizes Breast Cancer Cells to Bcl-2 Inhibitor, ABT-737, through Regulating Mcl-1. Radiat. Res. 2014, 182, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Hann, C.L.; Daniel, V.C.; Sugar, E.A.; Dobromilskaya, I.; Murphy, S.C.; Cope, L.; Lin, X.; Hierman, J.S.; Wilburn, D.L.; Watkins, D.N.; et al. Therapeutic Efficacy of ABT-737, a Selective Inhibitor of BCL-2, in Small Cell Lung Cancer. Cancer Res. 2008, 68, 2321–2328. [Google Scholar] [CrossRef] [Green Version]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R.; González-López, C.; Ou, H.L.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-Conjugation of Navitoclax as an Efficient Strategy to Increase Senolytic Specificity and Reduce Platelet Toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac Glycosides Are Broad-Spectrum Senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Fung, A.S.; Wu, L.; Tannock, I.F. Concurrent and Sequential Administration of Chemotherapy and the Mammalian Target of Rapamycin Inhibitor Temsirolimus in Human Cancer Cells and Xenografts. Clin. Cancer Res. 2009, 15, 5389–5395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and Exploiting Vulnerabilities for the Treatment of Liver Cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-Cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Kumarasamy, V.; Chung, S.; Ruiz, A.; Vail, P.; Tzetzo, S.; Wu, J.; Nambiar, R.; Sivinski, J.; Chauhan, S.S.; et al. Targeting Dual Signalling Pathways in Concert with Immune Checkpoints for the Treatment of Pancreatic Cancer. Gut 2021, 70, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.; Schörg, B.F.; Ahmetlić, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer Immune Control Needs Senescence Induction by Interferon-Dependent Cell Cycle Regulator Pathways in Tumours. Nat. Commun. 2020, 11, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T Cells Reverse Senescence-Associated Pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Agostini, A.; Mondragõn, L.; Bernardos, A.; Martínez-Máñez, R.; Dolores Marcos, M.; Sancenõn, F.; Soto, J.; Costero, A.; Manguan-García, C.; Perona, R.; et al. Targeted Cargo Delivery in Senescent Cells Using Capped Mesoporous Silica Nanoparticles. Angew. Chem. Int. Ed. 2012, 51, 10556–10560. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A Versatile Drug Delivery System Targeting Senescent Cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef]
- Thapa, R.K.; Nguyen, H.T.; Jeong, J.H.; Kim, J.R.; Choi, H.G.; Yong, C.S.; Kim, J.O. Progressive Slowdown/Prevention of Cellular Senescence by CD9-Targeted Delivery of Rapamycin Using Lactose-Wrapped Calcium Carbonate Nanoparticles. Sci. Rep. 2017, 7, 43299. [Google Scholar] [CrossRef] [Green Version]
- Ke, S.; Lai, Y.; Zhou, T.; Li, L.; Wang, Y.; Ren, L.; Ye, S. Molybdenum Disulfide Nanoparticles Resist Oxidative Stress-Mediated Impairment of Autophagic Flux and Mitigate Endothelial Cell Senescence and Angiogenic Dysfunctions. ACS Biomater. Sci. Eng. 2018, 4, 663–674. [Google Scholar] [CrossRef]
- Thoppil, H.; Riabowol, K. Senolytics: A Translational Bridge between Cellular Senescence and Organismal Aging. Front. Cell Dev. Biol. 2020, 7, 367. [Google Scholar] [CrossRef] [PubMed]
- Wissler Gerdes, E.O.; Misra, A.; Netto, J.M.E.; Tchkonia, T.; Kirkland, J.L. Strategies for Late Phase Preclinical and Early Clinical Trials of Senolytics. Mech. Ageing Dev. 2021, 200, 111591. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Senescent Cell Feature | Biomarker | Marker Level Change | Senescence Type | Detection Method |
|---|---|---|---|---|
| morphological changes | morphology, size | wide and flattened | general | light microscopy, FC |
| lysosomal activity | SA-β-galactosidase | increased | general | enzymatic staining |
| SA-α-fucosidase | increased | general | enzymatic staining | |
| Lipofuscin | increased | general | SBB, GL13 | |
| DNA damage, DDR | γH2AX | increased | general/damage-induced | IF |
| Mre11 | increased | general/damage-induced | IF | |
| Rad50 | increased | general/damage-induced | IF | |
| NSB1 | increased | general/damage-induced | IF | |
| ATM | increased | general/damage-induced | IF | |
| ATR | increased | general/damage-induced | IF | |
| 53BP1 | increased | general/damage-induced | IF | |
| MDC1 | increased | general/damage-induced | IF | |
| Rad17 | increased | general/damage-induced | IF | |
| TIF | increased | general/damage-induced | IF | |
| low/lack of DNA synthesis | BrdU | decreased | general | staining incorporation, IF |
| EdU | decreased | general | staining incorporation, IF | |
| lack of proliferation | Ki67 | decreased | general | IHC, IF |
| p16/pRB pathway | p16INK4a | increased | general | WB, IHC, IF |
| pRB | increased | general | WB, IHC, IF | |
| phospho-pRB | increased | general | WB, IHC, IF | |
| p53/p21 pathway | p53 | increased | general/damage-induced | WB, IHC, IF |
| p21 | increased | general/damage-induced | WB, IHC, IF | |
| phospho-p21 | increased | general/damage-induced | WB, IHC, IF | |
| DEC1 | increased | general/damage-induced | IHC, IF, special assay | |
| PPP1A | increased | general/damage-induced | IHC, special assay | |
| ROS | ROS | increased | general/oxidative stress-induced | fluorometry, FC |
| telomere length | telomere | decreased | replicative-induced | qPCR, FISH |
| SAHFs | SAHFs | increased | general/damage-induced | DAPI/Hoechst, confocal microscopy |
| HP1-gamma | increased | general/damage-induced | IF, IHC | |
| H3K9-methylation | increased | general/damage-induced | IF | |
| PML bodies | increased | general/damage-induced | IF | |
| nuclear membrane | lamin B1 | decreased | general | WB, IF, qPCR |
| cytokine secretion | SASPs | increased | damage-/oncogene-induced | WB, ELISA, SASP-assay |
| others | plasma membrane proteins | increased | general/replicative-/oncogene-induced | IF, WB, IHC, FC |
| apoptosis elimination | absent | general | IF, IHC |
| Term | Description |
|---|---|
| Senescence | Biological aging. Process of senescent cells (SCs) accumulation, SCs do not function, but they are metabolically active and remain in tissues. SCs are closely associated with age-related disorders. |
| Senotherapy (Senolysis) | Removal of senescent cells. |
| Senolytic drugs (Senolytics) | Class of drugs selectively eliminating SCs. |
| Senoblockers | Agents affecting epigenetic regulators to reactivate programs of youthfulness and regeneration. |
| Senomorphics | Small molecules inhibiting SASP. |
| Senostatics | Drugs interfering cells entering to senescence. |
| Senomodulators | Drugs suppressing SASP activity. |
| Senosuppressors | Therapeutics slowing down SCs accumulation rate. |
| Senolytic Drug Targets | Compound | Target | Note |
|---|---|---|---|
| Anti-apoptotic pathway | Navitoclax | BCL-2, BCL-XL, and BCL-w | ABT263 |
| ABT-737 | BCL-2, BCL-XL, and BCL-w | ABT263 paralogue and precursor | |
| A1331852 | BCL-XL | 2nd generation of BCL-2 family inhibitors | |
| A1155463 | BCL-XL | 2nd generation of BCL-2 family inhibitors | |
| Piperlongumine | apoptosis | An alkaloid, dietary natural product from Piper genus trees | |
| Geldanamycin | ? | Piperlongumine analogue | |
| Tanespimycin | ? | Piperlongumine analogue | |
| Alvespimycin | ? | Piperlongumine analogue | |
| Panobinostat | BCL-XL | increases 3/7 caspase activity | |
| PI3K and other kinases | Dasatinib | PI3K/Akt pathway | small molecule inhibiting various tyrosine kinases |
| Quercetin | PI3K/Akt and mTOR pathway | Flavonoid | |
| Fisetin | PI3K/Akt | natural flavonoid | |
| Luteolin | PI3K/Akt | Flavone | |
| Curcumin | PI3K/Akt | Flavone | |
| p53, p16, and DDR pathway | FOXO4-DRI | FOXO4 and p53 interaction | mitochondrial activity boost |
| 17-DMAG | HSP90/Akt | SASP suppressor | |
| KU-60019 | ATM | NF-κB inhibition | |
| NF-κB or C/EBPβ regulation | Rapamycin | mTORC1 complex | next generation Mdm2 inhibitor |
| Nutlin-3a | Mdm2 | p53 stabilization and SASP reduction | |
| MI-63 | Mdm2 | p53 stabilization and SASP reduction | |
| UBX0101 | Mdm2 | derived from Nutlin | |
| Metformin | SASP | NF-κB inhibition | |
| Resveratrol | SASP | NF-κB inhibition | |
| Ruxolitinib | JAK | INCB18424, C/EBPβ repression | |
| Momelotinib | JAK | CYT387, C/EBPβ repression | |
| Other | MCOPPB | NOP | anxiolytic opioid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int. J. Mol. Sci. 2022, 23, 4168. https://doi.org/10.3390/ijms23084168
Kudlova N, De Sanctis JB, Hajduch M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. International Journal of Molecular Sciences. 2022; 23(8):4168. https://doi.org/10.3390/ijms23084168
Chicago/Turabian StyleKudlova, Natalie, Juan Bautista De Sanctis, and Marian Hajduch. 2022. "Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs" International Journal of Molecular Sciences 23, no. 8: 4168. https://doi.org/10.3390/ijms23084168
APA StyleKudlova, N., De Sanctis, J. B., & Hajduch, M. (2022). Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. International Journal of Molecular Sciences, 23(8), 4168. https://doi.org/10.3390/ijms23084168