
Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
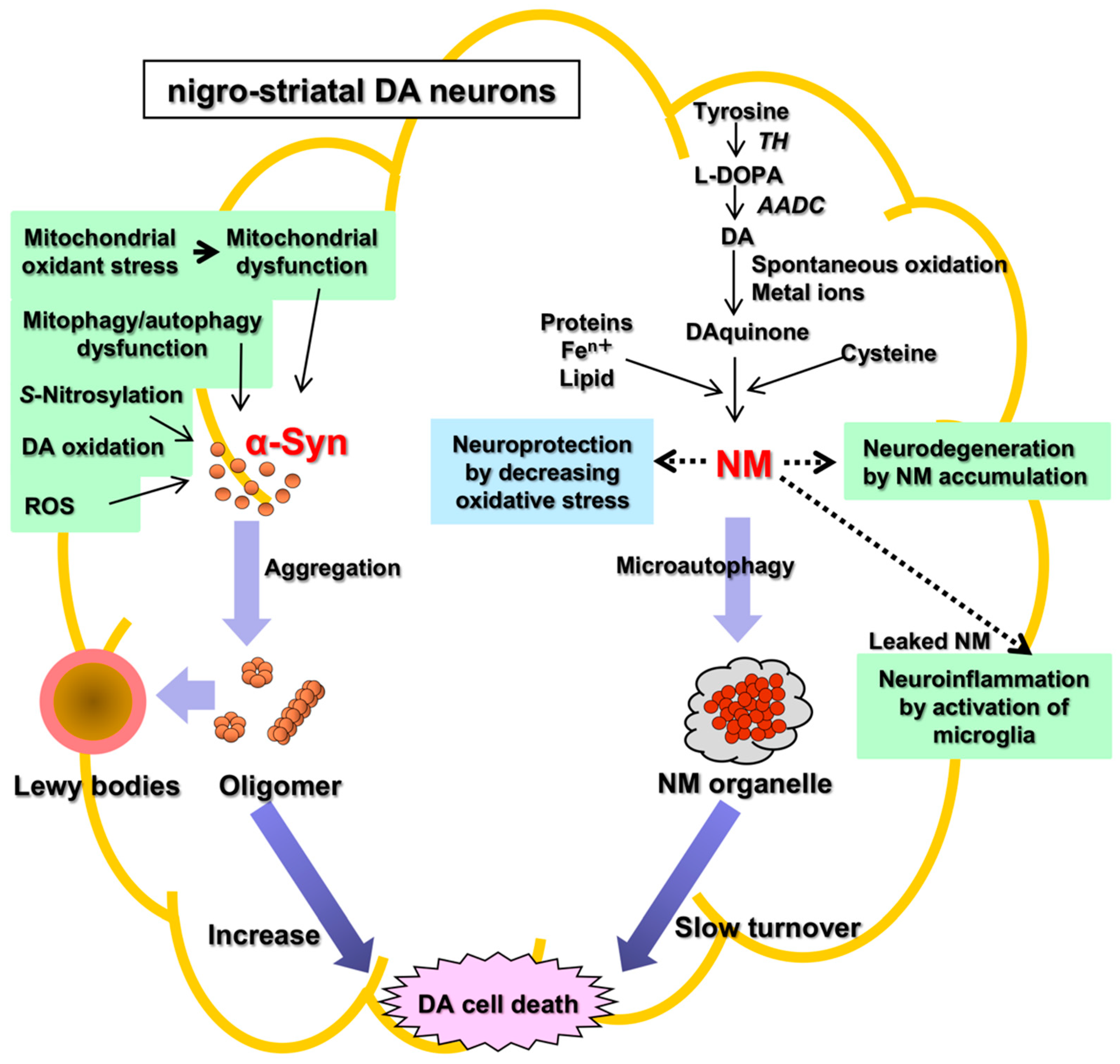
:1. Neuromelanin (NM) in Parkinson’s Disease
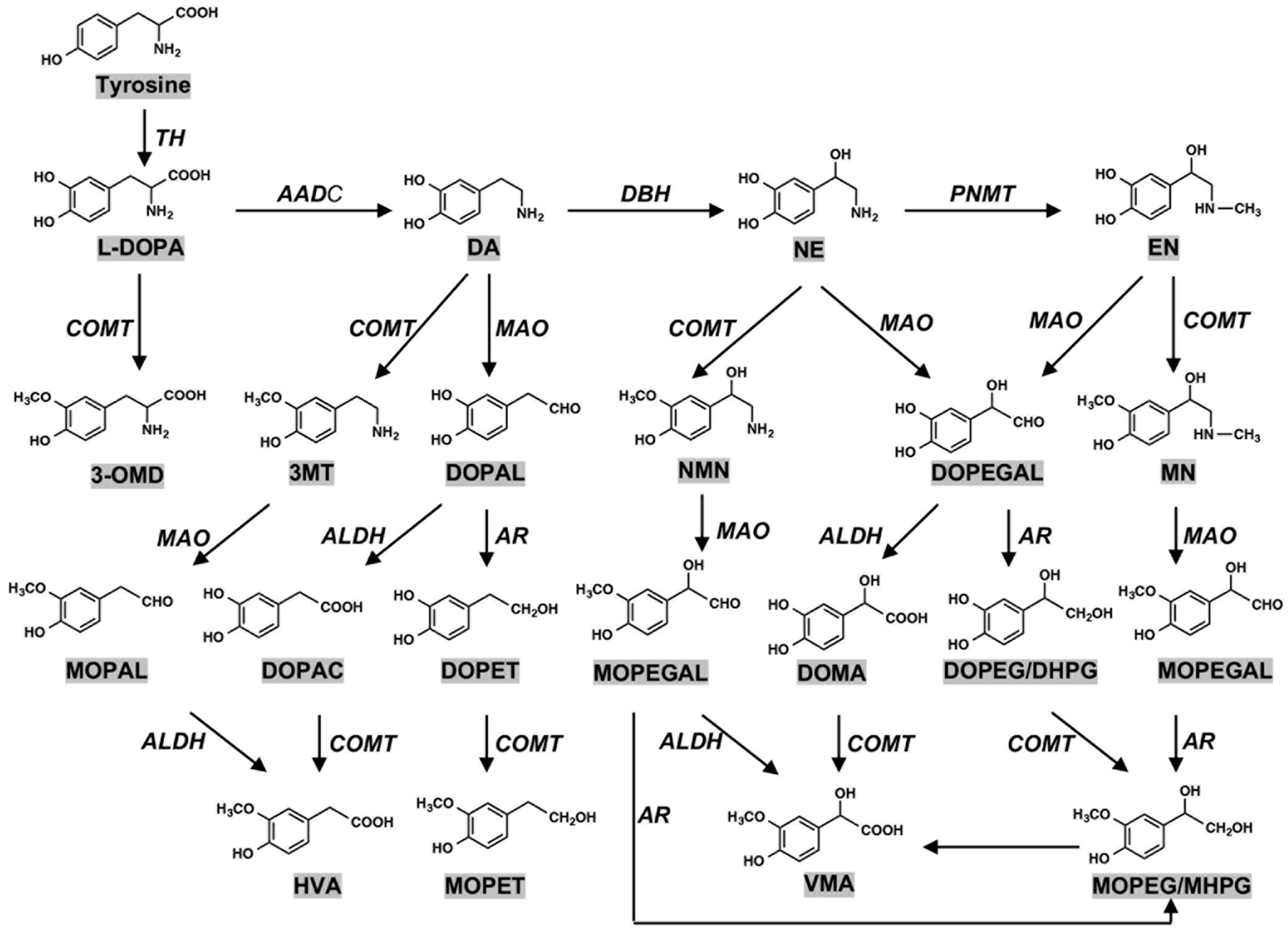
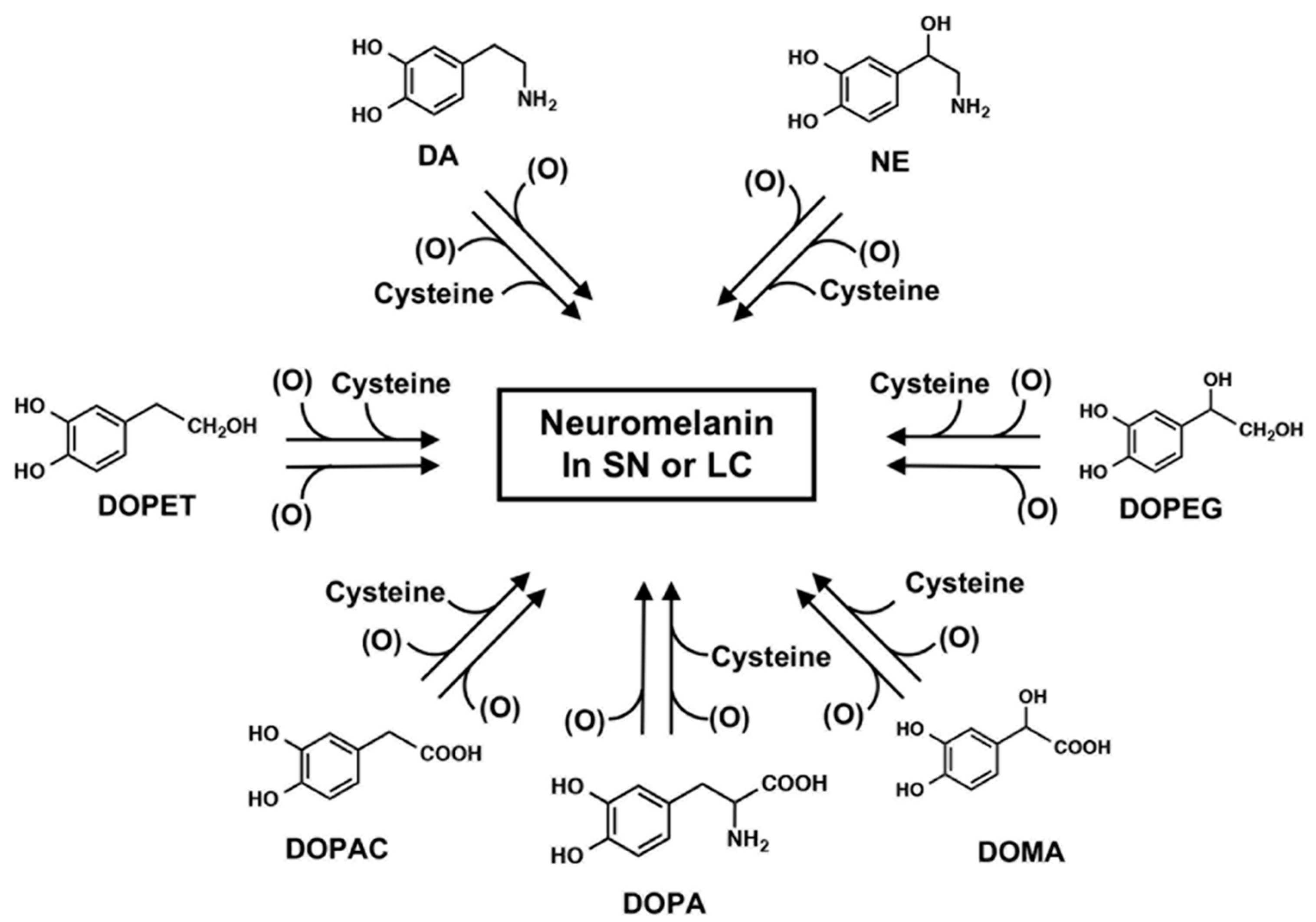
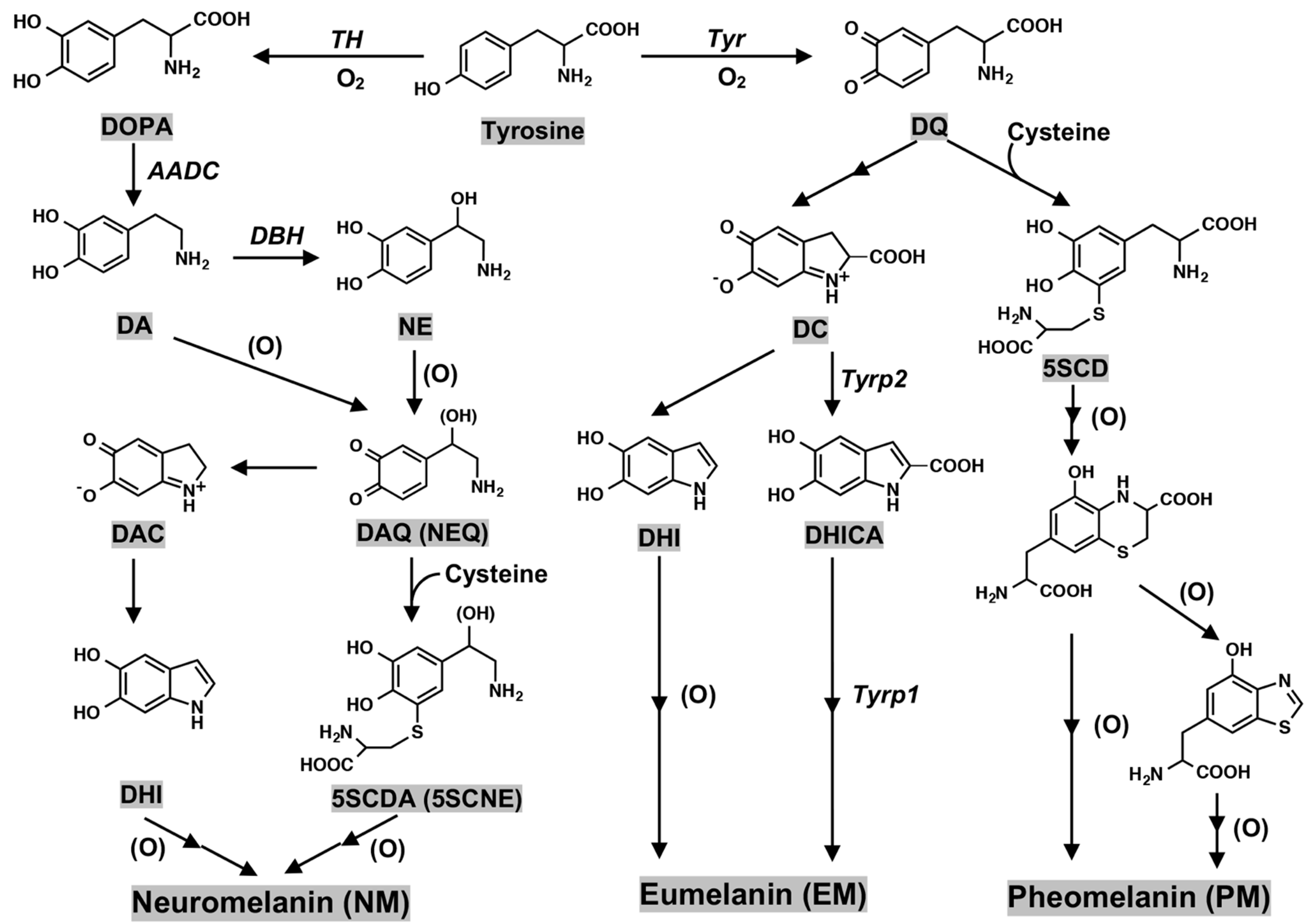
2. Biosynthesis of Neuromelanin (NM): Tyrosine Hydroxylase and Tyrosinase
3. Neuromelanin (NM): The Cause of Parkinson’s Disease?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AADC | aromatic L-amino acid decarboxylase |
| AAV | adeno-associated viral |
| ALDH | aldehyde dehydrogenase |
| AR | aldehyde reductase |
| BH4 | tetrahydrobiopterin |
| CD | cysteinyldopa |
| CDA | cysteinyldopamine |
| COMT | catechol-O-methyltransferase |
| DA | dopamine |
| DAC | dopaminechrome |
| DAQ | dopaminequinone |
| DBH | dopamine-β-hydroxylase |
| DC | dopachrome |
| DDC | dopa decarboxylase |
| DHI | 5,6-dihydroxyindole |
| DHICA | 5,6-dihydroxyindole-2-carboxylic acid |
| DQ | dopaquinone |
| DOMA | 3,4-dihydroxymandelic acid |
| DOPA | 3,4-dihydroxyphenylalanine |
| DOPAC DOPAL | 3,4-dihydroxyphenylacetic acid 3,4-dihydroxyphenylacetaldehyde |
| DOPEG | 3,4-dihydroxyphenylethylene glycol |
| DOPEGAL | 3,4-dihydroxyphenylglycolaldehyde |
| DOPET | 3,4-dihydroxyphenylethanol |
| EM | eumelanin |
| EN | epinephrine |
| euNM | eumelanic NM |
| HVA | homovanillic acid |
| LBD | Lewy body disease |
| LC | locus coeruleus |
| MAO | monoamine oxidase |
| MHPG | 3-methoxy-4-hydroxyphenylgycol |
| MN | metanephrine |
| MOPAL | 3-methoxy-4-hydroxyphenylacetaldehyde |
| MOPEG | 3-methoxy-4-hydroxyphenylethyleneglycol |
| MOPET | 3-methoxy-4-hydroxyphenylethanol |
| MOPEGAL | 3-methoxy-4-hydroxyphenylglycolaldehyde |
| MPTP | 1-phenyl-4-methyl-1,2,3,6-tetrahydropyridine |
| MSA | multiple system atrophy |
| NE | norepinephrine |
| NEQ | NEquinone |
| NM | neuromelanin |
| PD | Parkinson’s disease |
| fPD | familial Parkinson’s disease |
| sPD | sporadic Parkinson’s disease |
| pheoNM | pheomelanic NM |
| PNMT | phenylethanolamine N-methyltransferase |
| PM | pheomelanin |
| RBD | REM-sleep behavioral disorders |
| ROS | reactive oxygen species |
| 5SCD | 5-S-cysteinyldopa |
| 2SCD | 2-S-cysteinyldopa |
| SN | substantia nigra |
| SNCA | alpha-Synuclein gene |
| SNpc | substantia nigra pars compacta |
| TH | tyrosine hydroxylase |
| Tyr | tyrosinase |
| Tyrp1 | tyrosinase-related protein 1 |
| Tyrp2 | tyrosinase-related protein 2 |
| VMA | vanillylmandelic acid |
| VMAT-2 | vesicular monoamine transporter-2 |
References
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. Acad. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Guadagnolo, D.; Piane, M.; Torrisi, M.R.; Pizzuti, A.; Petrucci, S. Genotype-phenotype correlations in monogenic Parkinson disease: A review on clinical and molecular findings. Front. Neurol. 2021, 12, 648588. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.G.; Davidson, L.; Hornykiewicz, O. The neurochemistry of Parkinson’s disease: Effect of L-DOPA therapy. J. Pharmacol. Exp. Ther. 1975, 153, 453–464. [Google Scholar]
- Nagatsu, T.; Kato, T.; Numata (Sudo), Y.; Ikuta, K.; Sano, M.; Nagatsu, I.; Kondo, Y.; Inagaki, S.; Iizuka, R.; Hori, A.; et al. Phenylethanolamine N-methyltransferase and other enzymes of catecholamine metabolism in human brain. Clin. Chim. Acta 1977, 75, 221–232. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Biochemistry of postmortem brains in Parkinson’s disease: Historical overview and future prospects. J. Neural Transm. Suppl. 2007, 72, 113–120. [Google Scholar] [CrossRef]
- Fahn, S. The medical treatment of Parkinson disease from James Parkinson to George Cotzias. Mov. Disord. 2015, 30, 4–18. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. L-DOPA therapy for Parkinson’s disease: Past, present, and future. Parkinsonism. Relat. Disord. 2009, 15, S3–S8. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boye, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokoti, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, M.; Tadayon, R.; Salzano, G.; Shaw, G.S.; Walden, H. A mechanistic review of Parkin activation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129894. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signaling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRKK2 links to genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRKK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Buschauer, R.; Böhning, J.; Audagnotto, M.; Lasker, K.; Lu, T.-W.; Boassa, D.; Taylor, S.; Villa, E. The In Situ structure of Parkinson’s disease-linked LRRK2. Cell 2020, 182, 1508–1518. [Google Scholar] [CrossRef]
- Erb, M.L.; Moore, D.J. LRRK2 and endolysosomal system in Parkinson’s disease. J. Parkinson’s Dis. 2020, 10, 1271–1291. [Google Scholar] [CrossRef]
- Holdorff, B. Friedrich Heinrich Lewy (1885–1950) and his work. J. Hist. Neurosci. 2002, 11, 19–28. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Zecca, L.; Fariello, R.; Riederer, P.; Sulzer, D.; Gatti, A.; Tampellini, D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002, 510, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Barden, H. The histochemical distribution and localization of copper, iron, neuromelanin and lysosomal enzyme activity in the brain of aging rhesus monkey and the dog. J. Neuropathol. Exp. Neurol. 1971, 30, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Holdorff, B. Centenary of Tretiakoff’s thesis on the morphology of Parkinson’s disease, evolved on the grounds of encephalitis lethargica pathology. J. Hist. Neurosci. 2019, 28, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Fujikawa, K.; Zucca, F.A.; Zecca, L.; Ito, S. The structure of neuromelanin as studied by chemical degradative methods. J. Neurochem. 2003, 86, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Ohtara, K.; Ito, S. Chemical analysis of late stages of pheomelanogenesis: Conversion of dihydrobenzothiazine to a benzothiazole structure. Pigment Cell Melanoma Res. 2009, 22, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Murase, T.; Zucca, F.A.; Zecca, L.; Ito, S. Biosynthetic pathway to neuromelanin and its aging process. Pigment Cell Melanoma Res. 2012, 25, 792–803. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004, 56, 331–349. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Tanaka, H.; Tabuchi, K.; Ojika, M.; Zucca, F.A.; Zecca, L.; Ito, S. Reduction of the nitro group to amine by hydroiodic acid to synthesize o-aminophenol derivatives as putative degradative markers of neuromelanin. Molecules 2014, 19, 8039–8050. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Wakamatsu, K. Human hair melanins: What we have learned and have not learned from mouse coat color pigmentation. Pigment Cell Melanoma Res. 2011, 24, 63–74. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Tabuchi, K.; Ojika, M.; Zucca, F.A.; Zecca, L.; Ito, S. Norepinephrine and its metabolites are involved in the synthesis on neuromelanine derived from the locus coeruleus. J. Neurochem. 2015, 135, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raper, H.S. The tyrosinase-tyrosine reaction: Production from tyrosine of 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid—the precursors of melanin. Biochem. J. 1927, 21, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Mason, H.S. The chemistry of melanin; mechanism of the oxidation of dihydroxyphenylalanine by tyrosinase. J. Biol. Chem. 1948, 172, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ferrer, A.; Rodríguez-López, J.N.; García-Cánovas, F.; García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Cooksey, C.J.; Garratt, P.J.; Land, E.J.; Pavel, S.; Ramsden, C.A.; Riley, P.A.; Smit, N.P. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J. Biol. Chem. 1997, 272, 26226–26235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagatsu, T.; Levitt, M.; Udenfriend, S. Tyrosine hydroxylase: The initial step in norepinephrine biosynthesis. J. Biol. Chem. 1964, 239, 2910–2917. [Google Scholar] [CrossRef]
- Nagatsu, T.; Yamaguchi, T.; Rahman, M.R.; Trocewicz, J.; Oka, K.; Hirata, Y.; Nagatsu, I.; Narabayashi, H.; Kondo, K.; Iizuka, R. Catecholamine-related enzymes and the biopterin cofactor in Parkinson’s disease and related extrapyramidal diseases. Adv. Neurol. 1984, 40, 467–473. [Google Scholar] [PubMed]
- Nagatsu, T.; Nagatsu, I. Tyrosine hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other catecholamine-related enzymes, and their human genes in relation to the drug and gene therapy of Parkinson’s disease (PD): Historical overview and future prospects. J. Neural Transm. 2016, 123, 1255–1278. [Google Scholar] [CrossRef]
- Nagatsu, T.; Nakashima, A.; Ichinose, H.; Kobayashi, K. Human tyrosine hydroxylase in Parkinson’s disease and related disorders. J. Neural Transm. 2019, 126, 397–406. [Google Scholar] [CrossRef]
- Bose, A.; Petsko, G.A.; Eliezer, D. Parkinson’s disease and melanoma: Co-occurrence and mechanisms. J. Parkinson’s Dis. 2018, 8, 385–398. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wen, Y.; Al-Kuwari, N.; Chen, X. Association between Parkinson’s diseases and melanoma: Putting the pieces together. Front. Aging Neurosci. 2020, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, S.J.; Escott-Price, V.; Brice, A.; Gasser, T.; Pittman, A.M.; Bras, J.; Hardy, J.; Heutink, P.; Wood, N.M.; Singleton, A.B.; et al. Rare variants analysis of cutaneous malignant melanoma genes in Parkinson’s disease. Neurobiol. Aging 2016, 48, 222.e1–222.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leupold, D.; Szyc, L.; Stankovic, G.; Strobel, S.; Völker, H.-U.; Fleck, U.; Müller, T.; Scholz, L.; Riederer, P.; Monoranu, C.-M. Melanin and neuromelanin fluorescence studies focusing on Parkinson’s disease and its inherent risk for melanoma. Cells 2019, 8, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, M.; Baxter, D. On the nature of pigment granules in the cells of the locus coeruleus and substantia nigra. J. Neuropathol. Exp. Neurology 1958, 17, 586–598. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Jackson, I.J.; Urabe, K.; Montague, P.M.; Hearing, V.J. A second tyrosinase-related protein, TRP-2, is a melanogenic enzyme termed DOPAchrome tautomerase. EMBO J. 1992, 11, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Kroumpouzos, G.; Urabe, K.; Kobayashi, T.; Sakai, C.; Hearing, V.J. Functional analysis of the slaty gene product (TRP2) as dopachrome tautomerase and the effect of a point mutation on its catalytic function. Biochem. Biophys. Res. Commun. 1994, 202, 1060–1068. [Google Scholar] [CrossRef]
- Ancans, J.; Tobin, D.J.; Hoogduijn, M.J.; Smit, N.P.; Wakamatsu, K.; Thody, A.J. Melanosomal pH controls rate of melanogenesis, eumelanin/phaeomelanin ratio and melanosome maturation in melanocytes and melanoma cells. Exp. Cell Res. 2001, 268, 26–35. [Google Scholar] [CrossRef]
- Ito, S.; Suzuki, N.; Takebayashi, S.; Commo, S.; Wakamatsu, K. Neutral pH and copper ions promote eumelanogenesis after the dopachrome stage. Pigment Cell Melanoma Res. 2013, 26, 817–825. [Google Scholar] [CrossRef]
- Jiménez-Cervantes, C.; Solano, C.F.; Kobayashi, T.; Urabe, K.; Hearing, V.J.; Lozano, J.A.; García-Borrón, J.C. A new enzymatic function in the melanogenic pathway. The 5,6-dihydroxyindole-2-carboxylic acid oxidase activity of tyrosinase-related protein-1 (TRP1). J. Biol. Chem. 1994, 269, 17993–18000. [Google Scholar] [CrossRef]
- Olivares, C.; Jiménez-Cervantes, C.; Lozano, J.A.; Solano, F.; García-Borrón, J.C. The 5,6-dihydroxyindole-2-carboxylic acid (DHICA) oxidase activity of human tyrosinase. Biochem. J. 2001, 354, 131–139. [Google Scholar] [CrossRef]
- Boissy, R.E.; Sakai, C.; Zhao, H.; Kobayashi, T.; Hearing, V.J. Human tyrosinase-related protein-1 (TRP-1) does not function as a DHICA oxidase activity in contrast to murine TRP-1. Exp. Dermatol. 1998, 7, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Land, E.J.; Riley, P.A. Spontaneous redox reactions of dopaquinone and the balance between the eumelanic and phaeomelanic pathways. Pigment Cell Res. 2000, 13, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis-pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- D’ischia, M.; Wakamatsu, K.; Cicoira, F.; Mauro, E.D.; Garcia-Borron, J.C.; Commo, S.; Galván, I.; Ghanem, G.; Koike, K.; Meredith, P.; et al. Melanins and melanogenesis: From pigment cells to human health and technological applications. Pigment Cell Melanoma Res. 2015, 28, 520–544. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, K.; Zippin, J.H.; Ito, S. Chemical and biochemical control of skin pigmentation with special emphasis on mixed melanogenesis. Pigment Cell Melanoma Res. 2021, 34, 730–747. [Google Scholar] [CrossRef]
- Miranda, M.; Botti, D.; Bonfigli, A.; Ventura, T.; Acradi, A. Tyrosinase-like activity in normal human substantia nigra. Gen. Pharmacol. 1984, 15, 541–544. [Google Scholar] [CrossRef]
- Xu, Y.; Stokes, A.H.; Freeman, W.M.; Kumer, S.C.; Vogt, B.A.; Vrana, K.E. Tyrosinase mRNA is expressed in human substantia nigra. Mol. Brain Res. 1997, 45, 159–162. [Google Scholar] [CrossRef]
- Ikemoto, K.; Nagatsu, I.; Ito, S.; King, R.A.; Nishimura, A.; Nagatsu, T. Does tyrosinase exist in neuromelanin-pigmented neurons in the human substantia nigra? Neurosci. Lett. 1998, 253, 198–200. [Google Scholar] [CrossRef]
- Tief, K.; Schmidt, A.; Beermann, F. New evidence for tyrosinase in substantia nigra, forebrain and midbrain. Mol. Brain Res. 1998, 53, 307–310. [Google Scholar] [CrossRef]
- Greggio, E.; Bergantino, E.; Carter, D.; Ahmad, R.; Costin, G.-E.; Hearing, V.J.; Clarimon, J.; Singleton, A.; Eerola, J.; Hellström, O.; et al. Tyrosinase exacerbates dopamine toxicity but is not genetically associated with Parkinson’s disease. J. Neurochem. 2005, 93, 246–256. [Google Scholar] [CrossRef]
- Tribl, F.; Arzberger, T.; Riederer, P.; Gerlach, M. Tyrosinase is not detected in human catecholaminergic neurons by immunohistochemistry and Western blot analysis. J. Neural Transm. Suppl. 2007, 72, 51–55. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nakao, K.; Tanaka, H.; Kitahori, Y.; Tanaka, Y.; Ojika, M.; Ito, S. The oxidative pathway to dopamine-protein conjugates and their pro-oxidant activities: Implications for the neurodegeneration of Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.M.; Dryhurst, G. Iron- and manganese-catalyzed autoxidation of dopamine in the presence of L-cysteine: Possible insights into iron- and manganese-mediated dopaminergic neurotoxicity. Chem. Res. Toxicol. 1998, 11, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, oxidative stress and protein-quinone modifications in Parkinson’s and other neurodegenerative diseases. Angew. Chem. Int. Ed. Engl. 2019, 58, 6512–6527. [Google Scholar] [CrossRef]
- Ito, S.; Fujita, K. Conjugation of dopa and 5-S-cysteinyldopa with cysteine mediated by superoxide radical. Biochem. Pharmacol. 1982, 31, 2887–2889. [Google Scholar] [CrossRef]
- Ito, S. One-step synthesis of (2-amino-2-carboxyethylthio) dopas (cys-dopas) from dopa and cysteine by hydrogen peroxide in the presence of iron-EDTA complex. Bull. Chem. Soc. Jpn. 1983, 56, 365–366. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Fujita, K. Formation of cysteine conjugates from dihydroxyphenylalanine and its S-cysteinyl derivatives by peroxidase-catalyzed oxidation. Biochim. Biophys. Acta 1981, 672, 151–157. [Google Scholar] [CrossRef]
- Rosengren, E.; Linder-Eliasson, E.; Carlsson, A. Detection of 5-S-cysteinyldopamine in human brain. J. Neural Transm. 1985, 63, 247–253. [Google Scholar] [CrossRef]
- Ito, S.; Fujita, K.; Yoshioka, M.; Sienko, D.; Nagatsu, T. Identification of 5-S- and 2-S-cysteinyldopamine and 5-S-glutathionyldopamine formed from dopamine by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. 1986, 375, 134–140. [Google Scholar] [CrossRef]
- Fornstedt, B.; Pileblad, E.; Carlsson, A. In vivo autooxidation of dopamine in guinea pig striatum increases with age. J. Neurochem. 1990, 55, 578–586. [Google Scholar] [CrossRef]
- Haavic, J. L-DOPA is a substrate for tyrosine hydroxylase. J. Neurochem. 1977, 69, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Barek, H.; Zhao, H.; Heath, K.; Veraksa, A.; Sugumaran, M. Drosophila yellow-h encodes dopaminechrome tautomerase: A new enzyme in the eumelanin biosynthetic pathway. Pigment Cell Res. 2022, 35, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Badillo-Ramírez, I.; Saniger, J.M.; Rivas-Arancibia, S. 5-S-cysteinyl-dopamine, a neurotoxic endogenous metabolite of dopamine: Implications for Parkinson’s disease. Neurochem. Int. 2019, 129, 104514. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.D.; Garguilo, J.; Zucca, F.A.; Albertini, A.; Zecca, L.; Edwards, G.S.; Nemanich, R.J.; Simon, J.D. The surface oxidation potential of human neuromelanin reveals a spherical architecture structure with a pheomelanin core and a eumelanin surface. Proc. Nat. Acad. Sci. USA 2006, 103, 14785–14789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzer, D.; Bogulavsky, J.; Larsen, K.E.; Behr, G.; Karatekin, E.; Kleinman, M.H.; Turro, N.; Krantz, D.; Edwards, R.H.; Greene, L.A.; et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Nat. Acad. Sci. USA 2000, 97, 11869–11874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecca, L.; Zucca, F.A.; Costi, P.; Tampellini, D.; Gatti, A.; Gerlach, M.; Riederer, P.; Fariello, R.G.; Ito, S.; Gallorini, M.; et al. The neuromelanin of human substantia nigra: Structure, synthesis and molecular behaviour. J. Neural Transm. Suppl. 2003, 65, 145–155. [Google Scholar] [CrossRef]
- Plum, S.; Steinbach, S.; Attems, J.; Keers, S.; Riederer, P.; Gerlach, M.; May, C.; Marcus, K. Proteomic characterization of neuromelanin granules isolated from human substantia nigra by laser-microdissection. Sci. Rep. 2016, 6, 37139. [Google Scholar] [CrossRef] [Green Version]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Paris, I.; Muňoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Tribl, F.; Marcus, K.; Meyer, H.E.; Bringmann, G.; Gerlach, M.; Riederer, P. Subcellular proteomics reveals neuromelanin granules to be lysosome-related organelle. J. Neural Transm. 2006, 113, 741–749. [Google Scholar] [CrossRef]
- Zucca, F.A.; Vanna, R.; Cupaioli, F.A.; Bellei, C.; De Palma, A.; Di Silvestre, D.; Mauri, P.; Grassi, S.; Prinetti, A.; Casella, L.; et al. Neuromelanin organelles are specialized autophagosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Parkinson’s Dis. 2018, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Ohta, S.; Tanaka, S.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Reichmann, H.; Riederer, P. Biochemische Analyse der Atmungskettenkomplex verschiedener Hirnregionen von Patienten mit M. Parkinson. In Proceedings of the Symposium zu Morbus Parkinson und andere Basalganglienerkrankungen, Bad Kissingen, Germany, 23–25 April 1989; Ministerium für Forschung und Technologie (BMBF): Bad Kissingen, Germany, 1989; p. 44. [Google Scholar]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gaol, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Hattori, N.; Saiki, S.; Imai, Y. Regulation by mitophagy. Int. J. Biochem. Cell Biol. 2014, 53, 147–150. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease; from mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Riederer, P. The nigral coup in Parkinson’s disease by α-synuclein and its associated rebels. Cells 2021, 10, 598. [Google Scholar] [CrossRef]
- Wright, R. Mitochondrial dysfunction and Parkinson’s disease. Nat. Neurosci. 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Cook, C.; Petrucelli, L. A Critical evaluation of the ubiquitin-proteasome system in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.K.; Ludtman, M.H.R.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-synuclein and complex I deficiency: Explorations of their relationship in differentiated neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef] [PubMed]
- Mor, D.E.; Tsika, E.; Mazzulli, J.R.; Gould, N.S.; Kim, H.; Daniels, M.J.; Doshi, S.; Gupta, P.; Grossman, J.L.; Tan, V.X.; et al. Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nature Neurosci. 2017, 20, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wang, Y.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.F.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar α-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020, 295, 17497–17513. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Aging Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef] [PubMed]
- Grünblatt, E.; Riederer, P. Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. J. Neural Transm. 2016, 123, 83–90. [Google Scholar] [CrossRef]
- Goldsein, D.S. The catecholaldehyde hypothesis: Where MAO fits in. J. Neural Transm. 2020, 127, 169–177. [Google Scholar] [CrossRef]
- Ito, S.; Tanaka, H.; Ojika, M.; Wakamatsu, K.; Sugumaran, M. Oxidative transformations of 3,4-dihydroxyphenylacetaldehyde generate potential reactive intermediates as causative agents for its neurotoxicity. Int. J. Mol. Sci. 2021, 22, 11751. [Google Scholar] [CrossRef]
- Sahay, S.; Ghosh, D.; Singh, P.K.; Maji, S.K. Alteration of structure and aggregation of α-synuclein by familial Parkinson’s disease associated mutations. Curr. Protein Pept. Sci. 2017, 18, 656–676. [Google Scholar] [CrossRef]
- Double, K.L.; Ben-Shachar, D.; Youdim, M.B.H.; Zecca, L.; Riederer, P.; Gerlach, M. Influence of neuromelanin on oxidative pathways within the human substantia nigra. Neurotoxicol. Teratol. 2002, 24, 621–628. [Google Scholar] [CrossRef]
- McCann, H.; Cartwright, H.; Halliday, G.M. Neuropathology of α-synuclein progression and Braak hypothesis. Mov. Disord. 2016, 31, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.-M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.-J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1-/- mice. Nature 2019, 571, 569. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, Y.; Wang, W.; Wang, Y.; Liu, H.; Liu, F.; Chen, R.; Dawson, V.L.; Dawson, T.M.; Lu, F.; et al. Molecular mediation of prion-like α-synuclein fibrillation from toxic PFFs to nontoxic species. ACS App. Biol. Mater. 2020, 3, 6096–6102. [Google Scholar] [CrossRef]
- Riederer, P.; Berg, D.; Casadei, N.; Cheng, F.; Classen, J.; Dresel, C.; Jost, W.; Krüger, R.; Müller, T.; Reichmann, H.; et al. α-Synuclein in Parkinson’s disease: Causal or bystanders? J. Neural. Transm. 2019, 126, 815–840. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Oh, C.-K.; Dolatabadi, N.; Cieplak, P.; Diaz-Meco, M.T.; Moscat, J.; Nolan, J.P.; Nakashima, T.; Lipton, S.A. S-Nitrosylation of p62 inhibits autophagic flux to promote α-synuclein secretion and spread in Parkinson’s disease and Lewy body dementia (LBD). J. Neurosci. 2022, 42, 3011–3024. [Google Scholar] [CrossRef]
- Meade, R.M.; Watt, K.J.C.; Williams, R.J.; Mason, J.M. A downsized and optimized intracellular library-derived peptide prevents alpha-synuclein primary nucleation and toxicity without impacting upon lipid binding. J. Mol. Biol. 2021, 433, 167323. [Google Scholar] [CrossRef]
- Taguchi, T.; Ikuno, M.; Hondo, M.; Parajuli, L.K.; Taguchi, K.; Ueda, J.; Sawamura, M.; Okuda, S.; Nakanishi, E.; Hara, J.; et al. α-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: A prodromal Parkinson’s disease model. Brain 2020, 143, 249–265. [Google Scholar] [CrossRef]
- Blesa, J.; Foffani, G.; Dehay, B.; Bezard, E.; Obeso, J.A. Motor and non-motor circuits disturbances in early Parkinson disease: Which happen first? Nat. Rev. Neurosci. 2022, 23, 115–128. [Google Scholar] [CrossRef]
- Götz, M.E.; Doulble, K.; Gerlach, M.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hülsmann, J. Iron as the concert master in the pathogenic orchestra playing in sporadic Parkinson’s disease. J. Neural. Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. Iron in brain function and dysfunction with emphasis on Parkinson’s disease. Eur. Neurol. 1991, 31, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Double, K.L.; Ben-Shachar, D.; Zecca, L.; Youdim, M.B.H.; Riederer, P. Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson’s disease. Neurotox. Res. 2003, 5, 35–44. [Google Scholar] [CrossRef]
- Mochizuki, H.; Choong, C.-J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm. 2020, 127, 181–187. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interaction of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Giménez, J.; Cuadros, T.; Bové, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Peñuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [Green Version]
- Vila, M. Neuromelanin, aging, and neuronal vulnerability in Parkinson’s disease. Mov. Disord. 2019, 34, 1440–1451. [Google Scholar] [CrossRef]
- Vila, M.; Laguna, A.; Carballo-Carbajal, I. Intracellular crowding by age-dependent neuromelanin accumulation disrupts neuronal proteostasis and triggers Parkinson disease pathology. Autophagy 2019, 15, 2028–2030. [Google Scholar] [CrossRef] [Green Version]
- Zucca, F.A.; Giaveri, G.; Gallorini, M.; Albertini, A.; Toscani, M.; Pezzoli, G.; Lucius, R.; Wilms, H.; Sulzer, D.; Ito, S.; et al. The neuromelanin of human substantia nigra: Physiological and pathogenic aspects. Pigment Cell Res. 2004, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-S.; Lin, K.-L.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Chan, P. Interaction between neuromelanin and alpha-synuclein in Parkinson’s disease. Biomolecules 2015, 5, 1122–1142. [Google Scholar] [CrossRef] [PubMed]
- Tessari, I.; Bisaglia, M.; Valle, F.; Samori, B.; Bergantino, E.; Mammi, S.; Bubacco, L. The reaction of α-synuclein with tyrosinase: Possible implication for Parkinson’s disease. J. Biol. Chem. 2008, 283, 16808–16817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Zhu, J.; Hwu, W.-J.; Jankovic, J. The role of alpha-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells. PLoS ONE 2012, 7, e45183. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, M.; Calero, O. The neuromelanin paradox and its dual role in oxidative stress and neurodegeneration. Antioxidants 2021, 10, 124. [Google Scholar] [CrossRef]
- Cebrian, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsatell, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Diederich, N.J.; Surmeier, D.J.; Uchihara, T.; Grillner, S.; Goetz, C.G. Parkinson’s disease: Is it a consequence of human brain evolution? Mov. Disord. 2019, 34, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Foffani, G.; Obeso, J.A. A cortical pathogenic theory of Parkinson’s disease. Neuron 2018, 99, 1116–1128. [Google Scholar] [CrossRef] [Green Version]
- Diederich, N.J.; Uchihara, T.; Grillner, S.; Goetz, C.G. The evolution-driven signature of Parkinson’s disease. Trends Neurosci. 2020, 43, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Cui, L.; Gao, J.; Zhu, M.; Zhang, Y.; Zhang, H.L. Gut microbial metabolites in Parkinson’s disease: Implications of mitochondrial dysfunction in the pathogenesis and treatment. Mol. Neurobiol. 2021, 58, 3745–3758. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 1–17. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagatsu, T.; Nakashima, A.; Watanabe, H.; Ito, S.; Wakamatsu, K. Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. Int. J. Mol. Sci. 2022, 23, 4176. https://doi.org/10.3390/ijms23084176
Nagatsu T, Nakashima A, Watanabe H, Ito S, Wakamatsu K. Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. International Journal of Molecular Sciences. 2022; 23(8):4176. https://doi.org/10.3390/ijms23084176
Chicago/Turabian StyleNagatsu, Toshiharu, Akira Nakashima, Hirohisa Watanabe, Shosuke Ito, and Kazumasa Wakamatsu. 2022. "Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase" International Journal of Molecular Sciences 23, no. 8: 4176. https://doi.org/10.3390/ijms23084176
APA StyleNagatsu, T., Nakashima, A., Watanabe, H., Ito, S., & Wakamatsu, K. (2022). Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. International Journal of Molecular Sciences, 23(8), 4176. https://doi.org/10.3390/ijms23084176