
The Tumor Microenvironment Drives Intrahepatic Cholangiocarcinoma Progression

 ,
,  , ,
, , 
 ,
,  and
and 
Abstract
:1. Introduction
2. Results
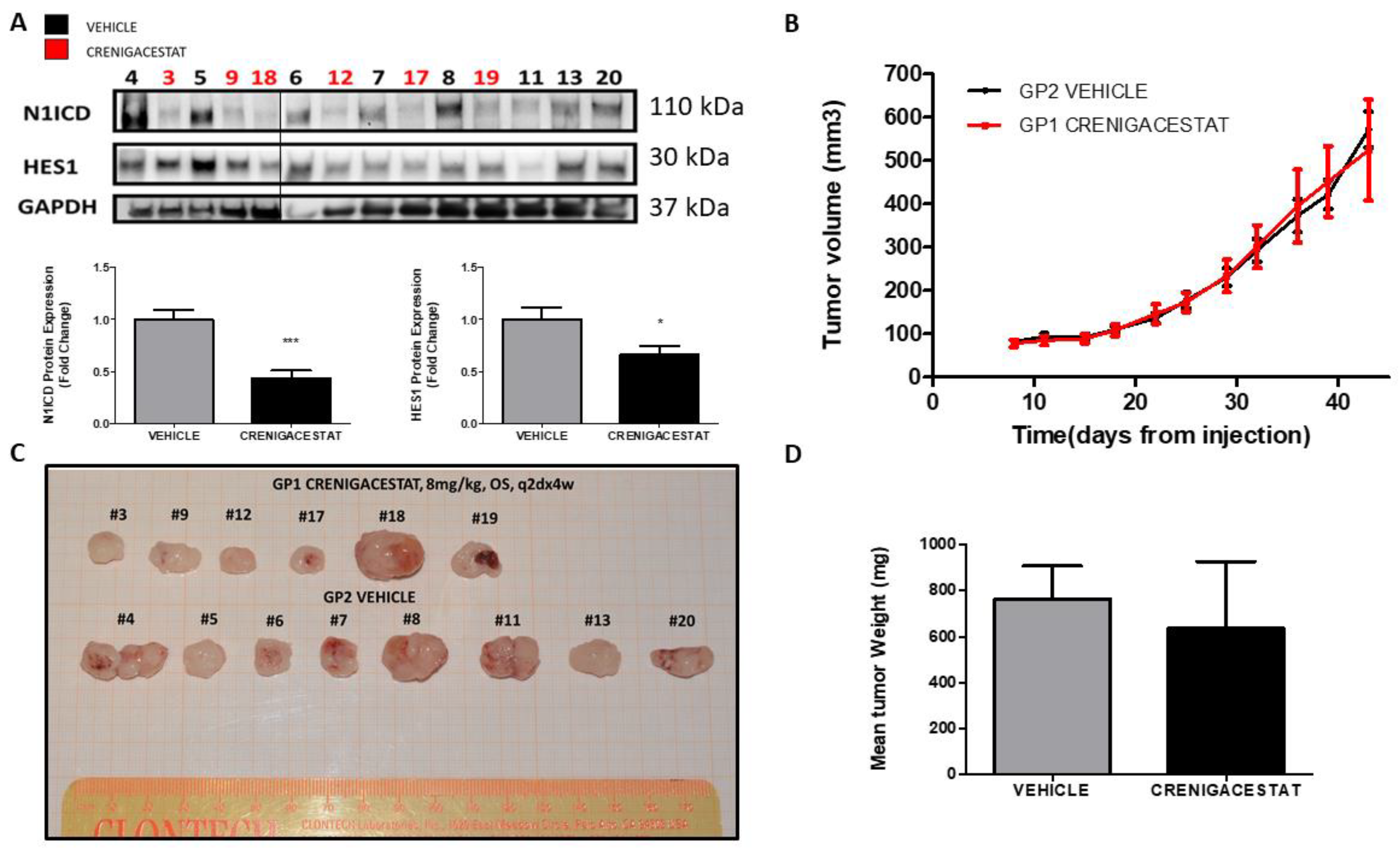
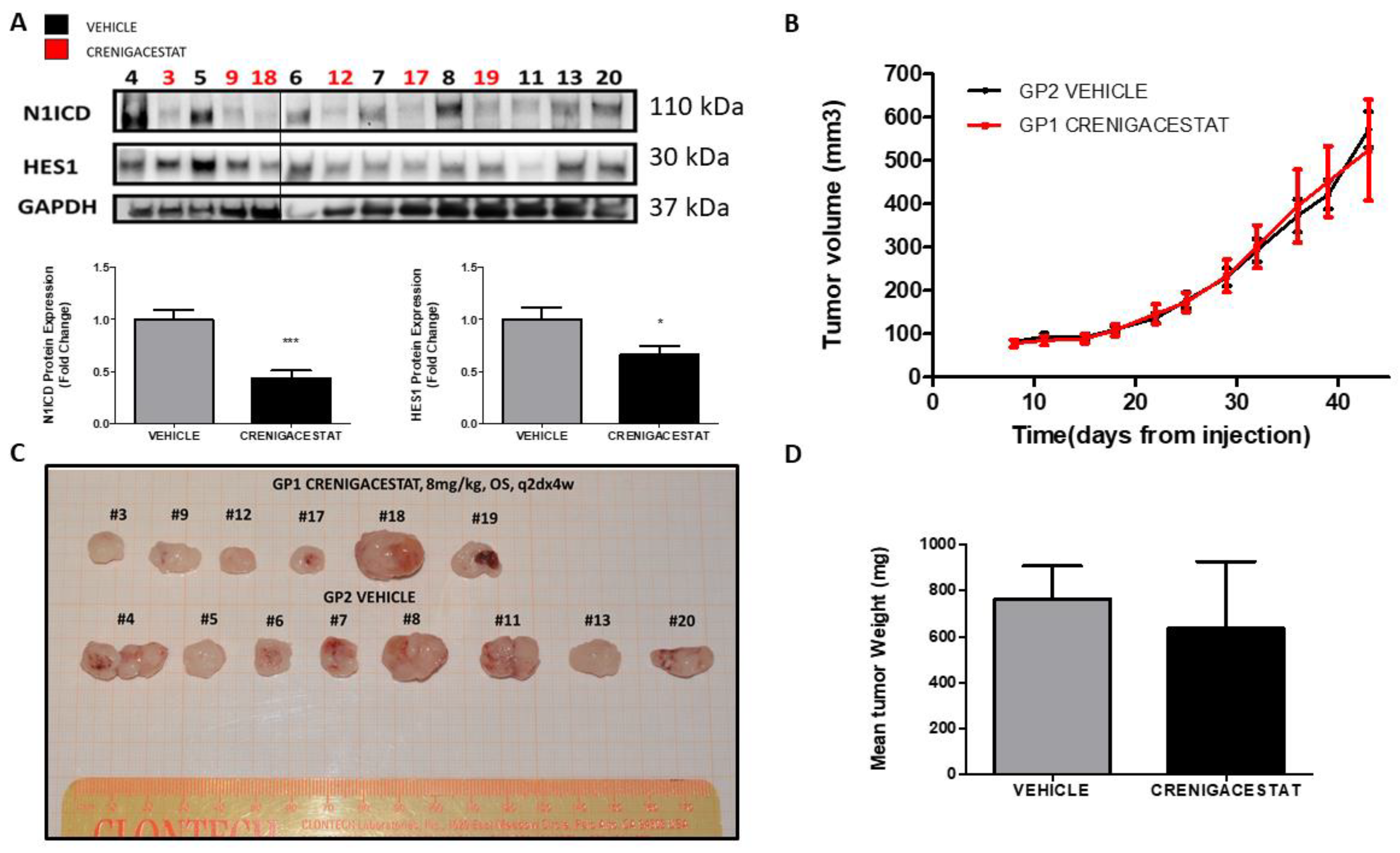
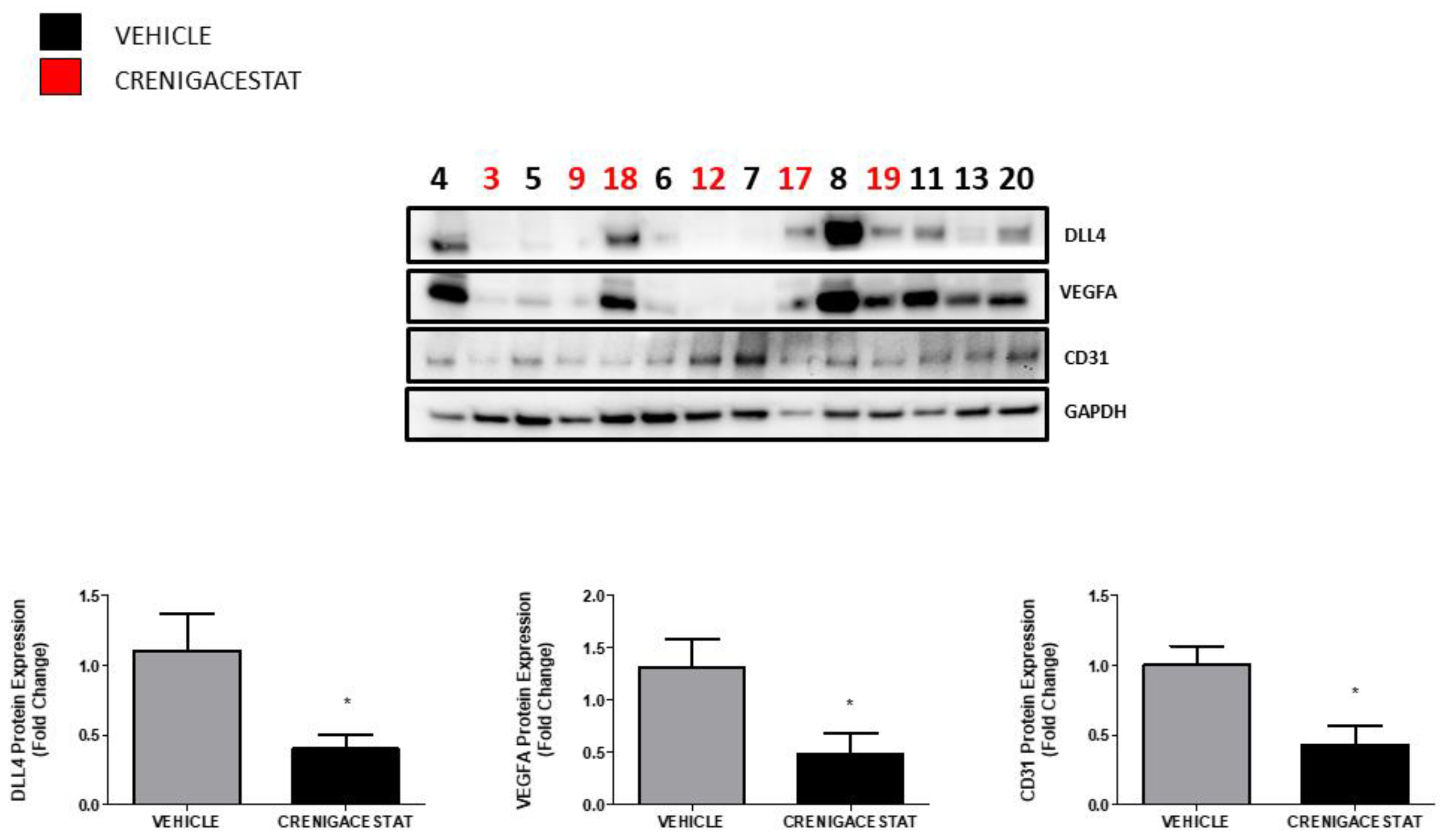
2.1. Crenigacestat Treatment in the HuCCT1-Xenograft Mouse Model
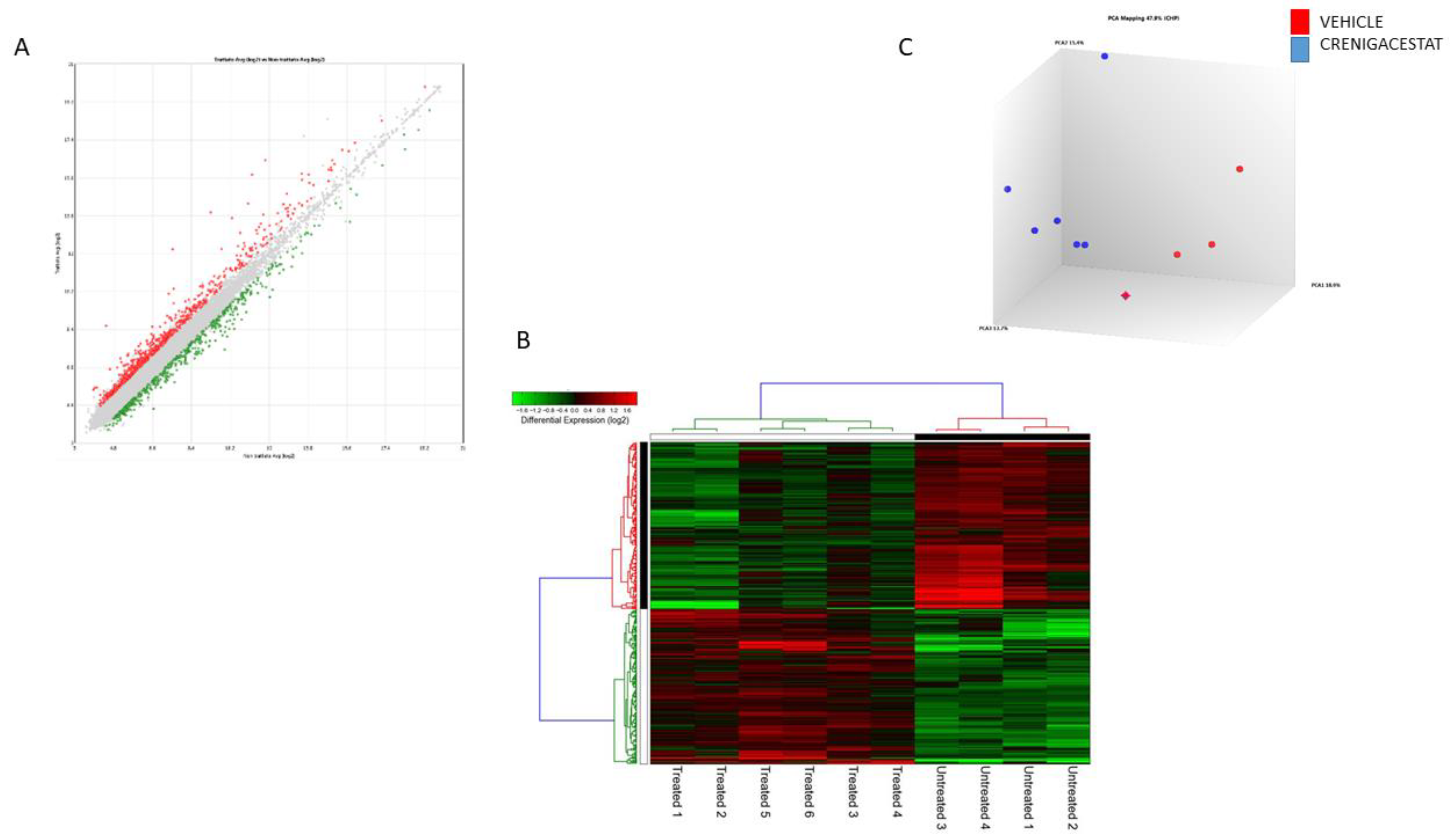
2.2. Gene Expression Analysis of the HuCCT1-Xenograft Mouse Model
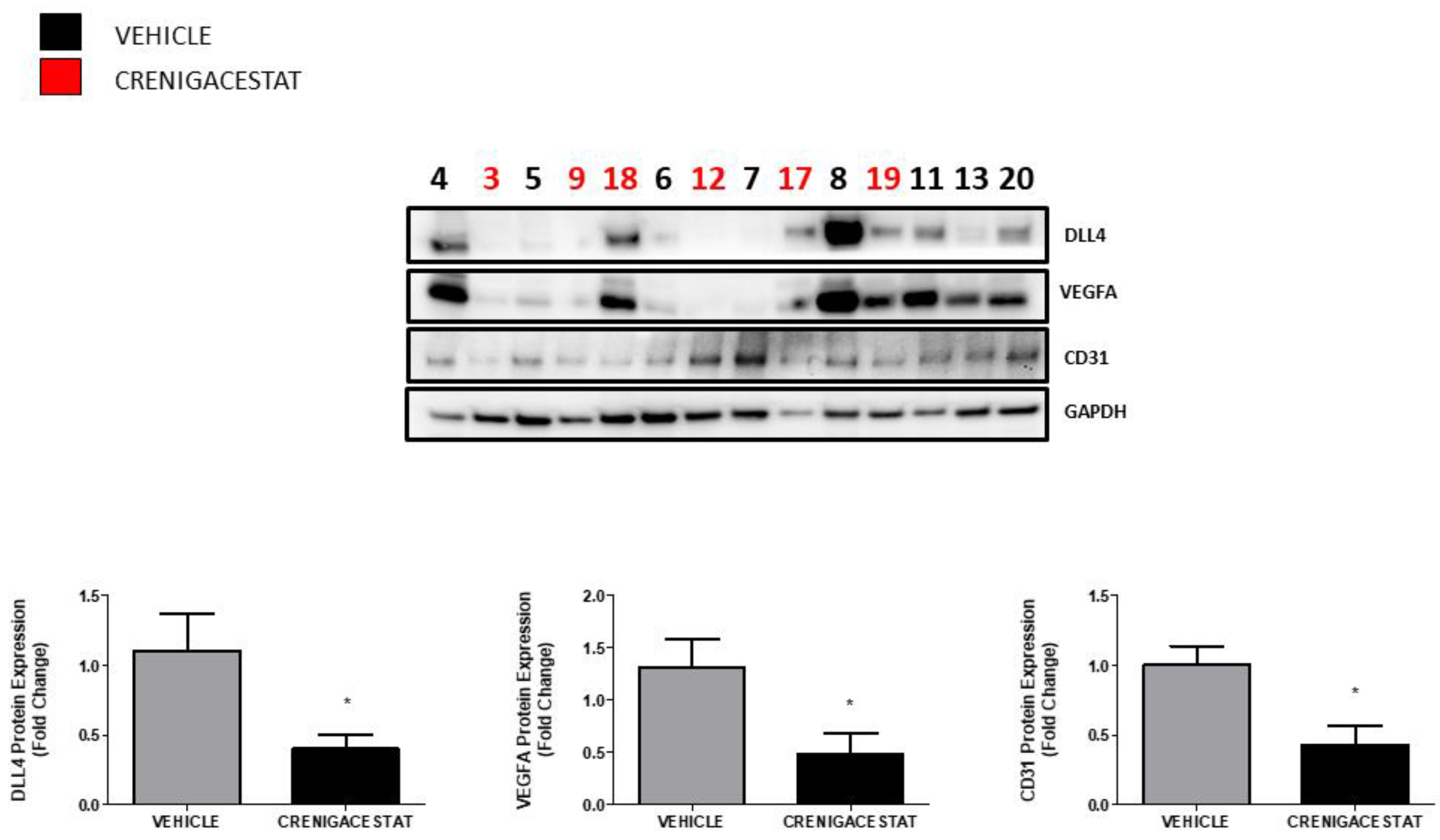
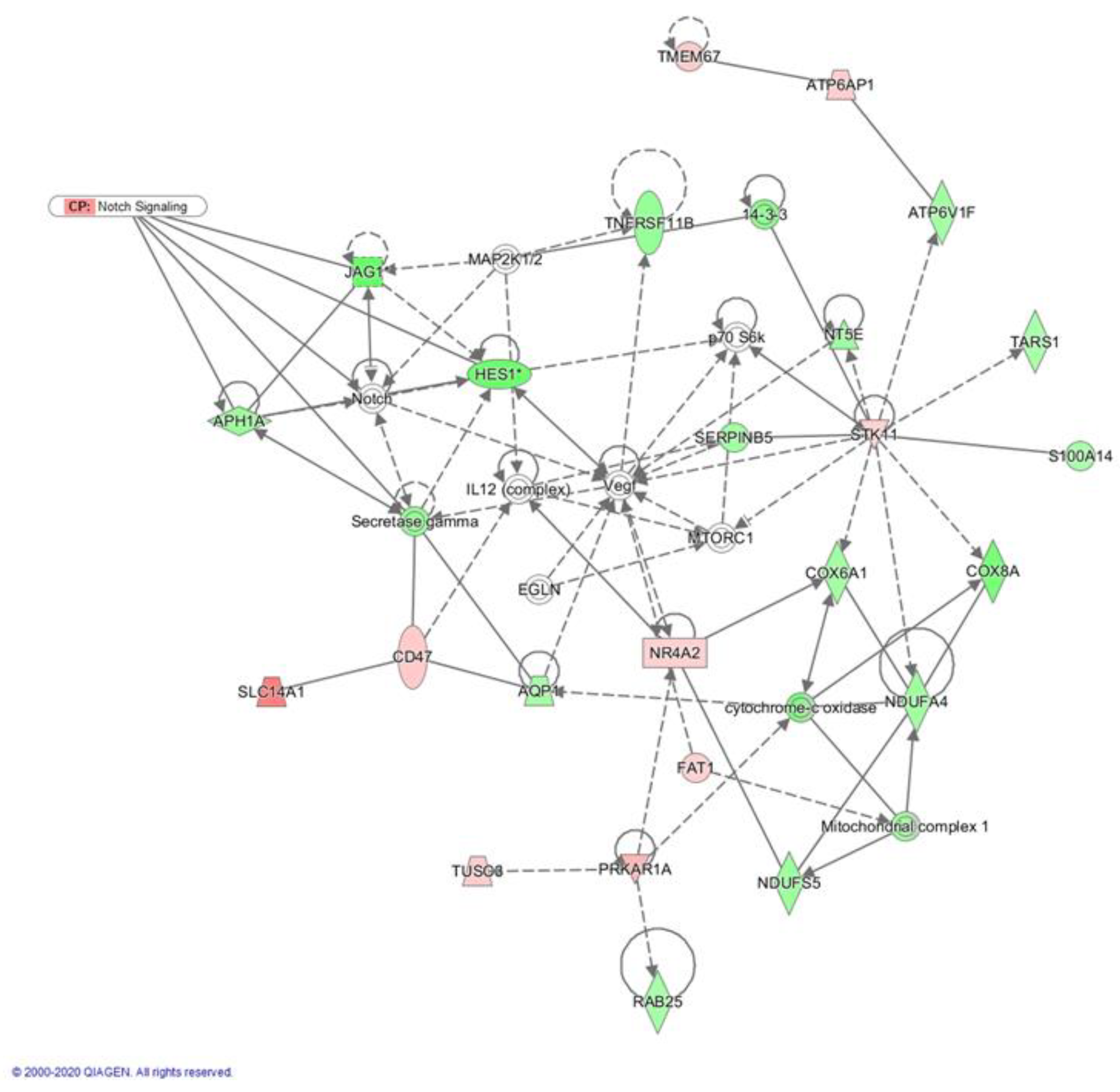
2.3. Crenigacestat Affects the Notch Pathway in the HuCCT1-Xenograft Mice Model
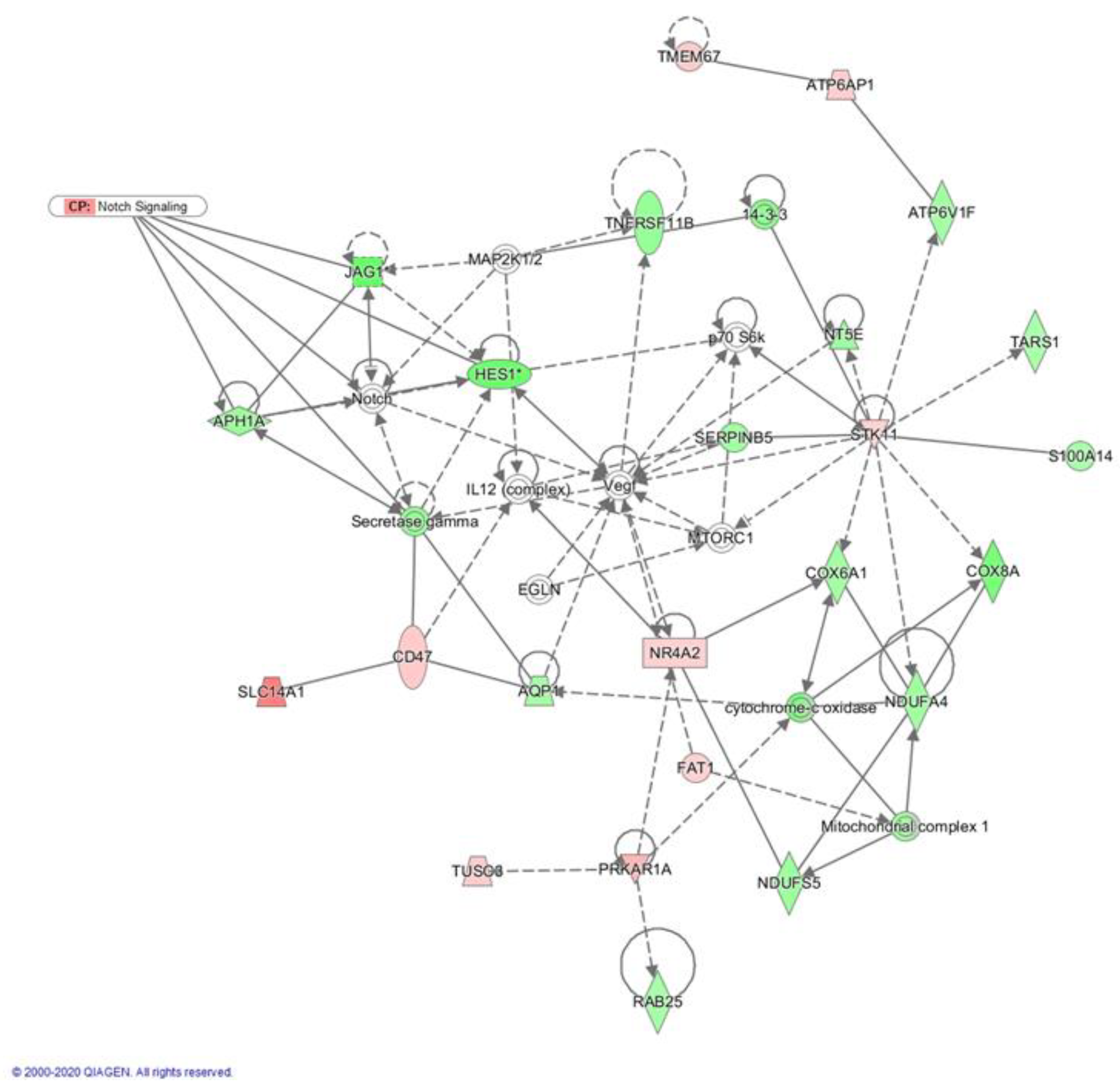
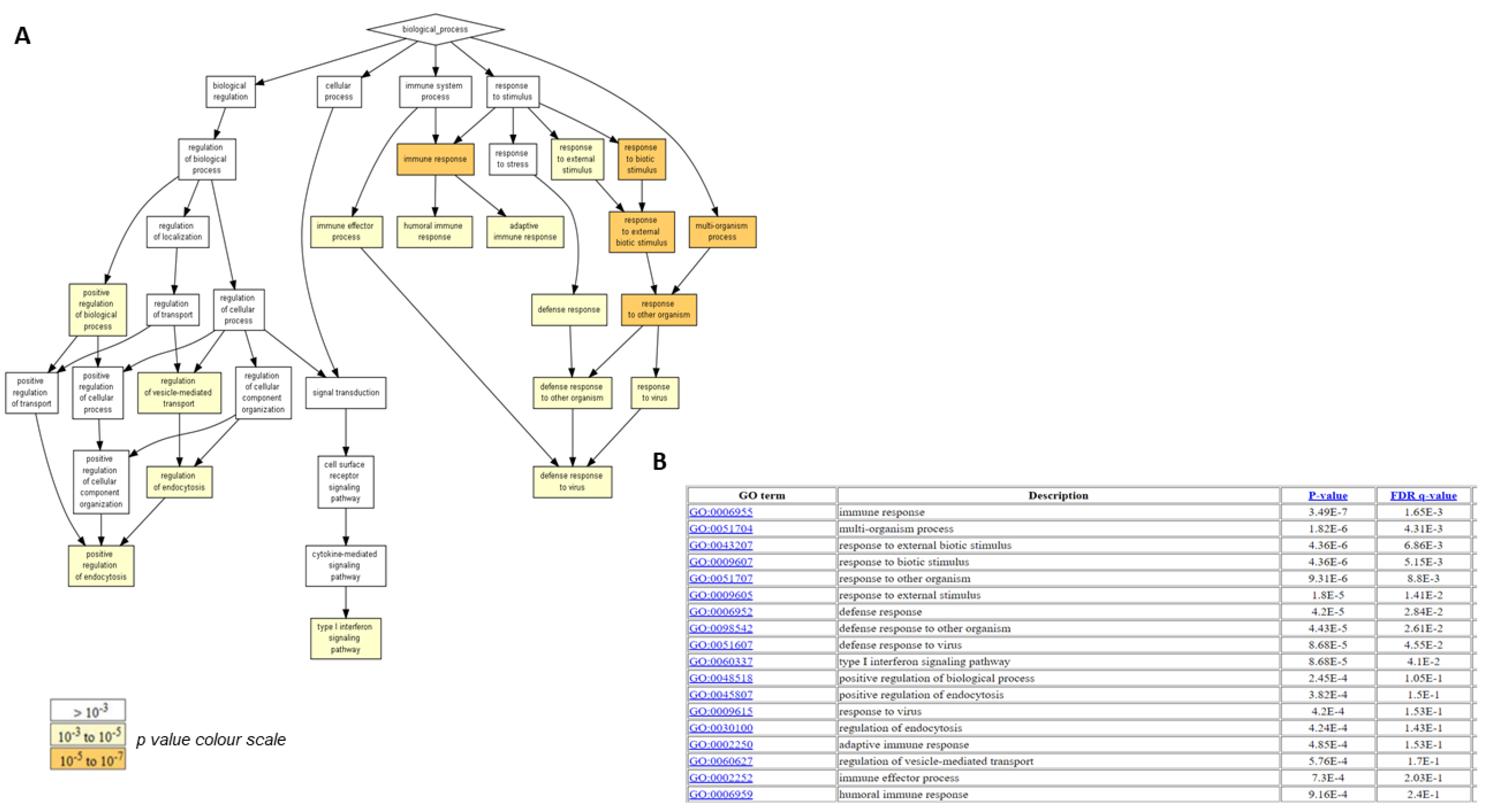
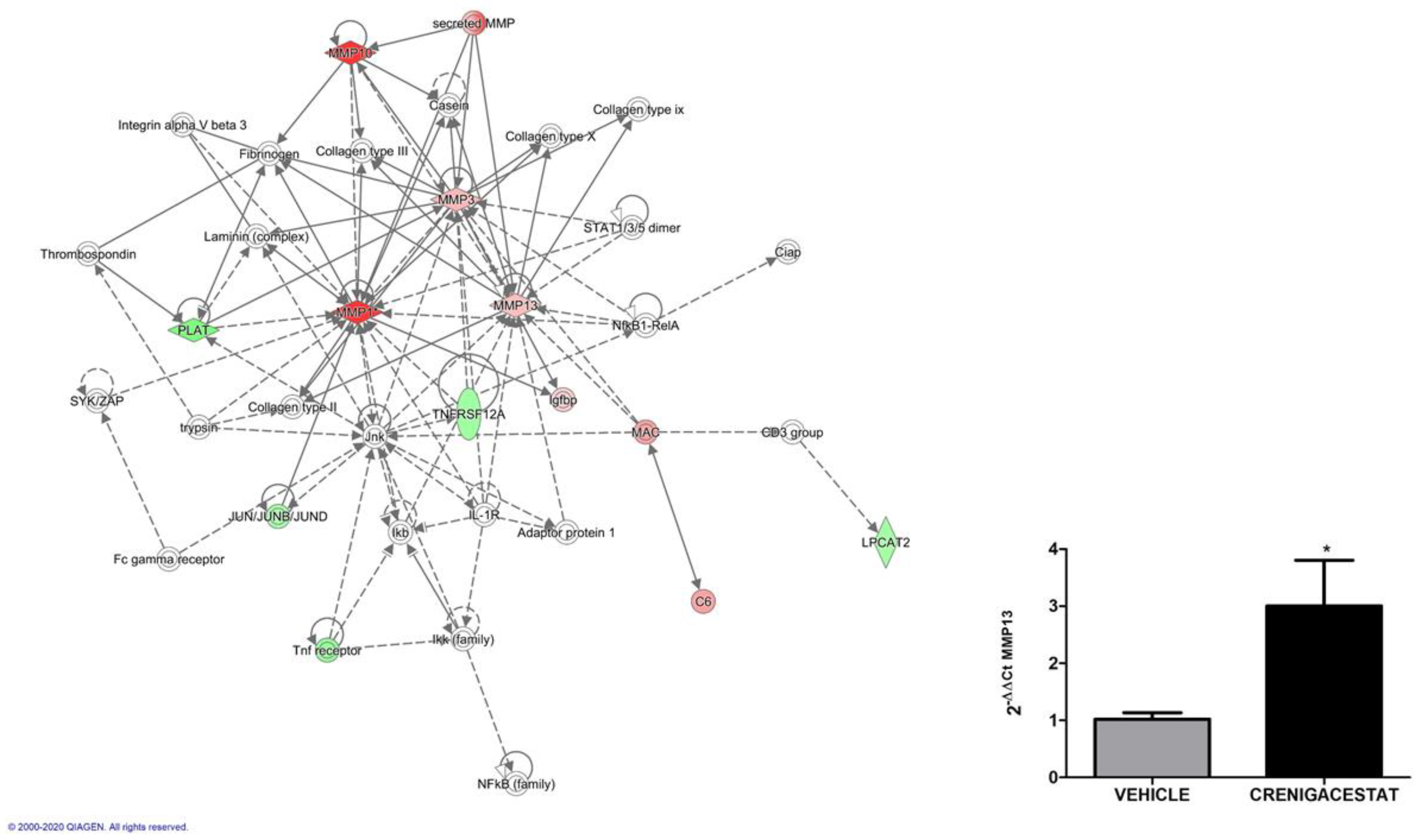
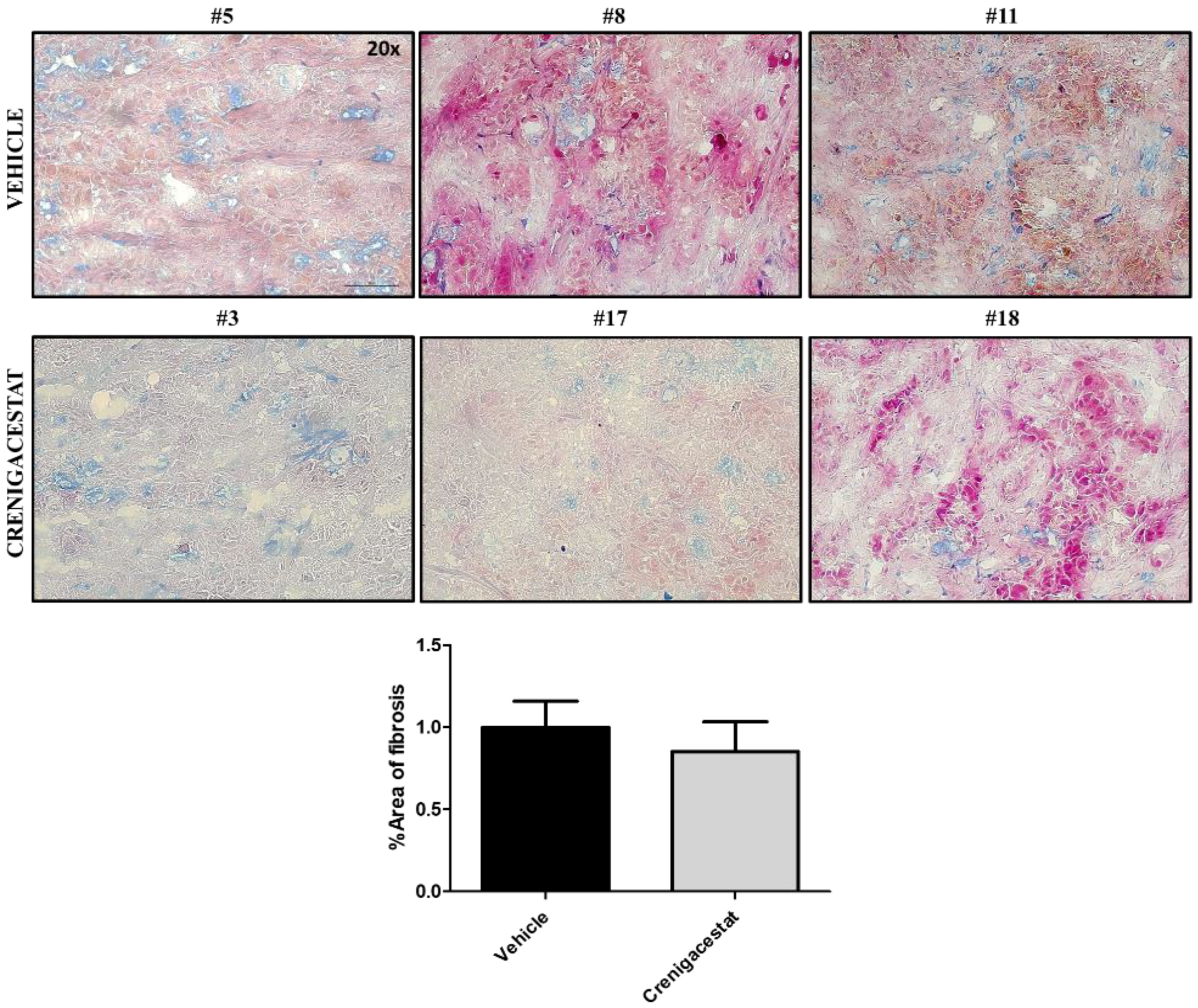
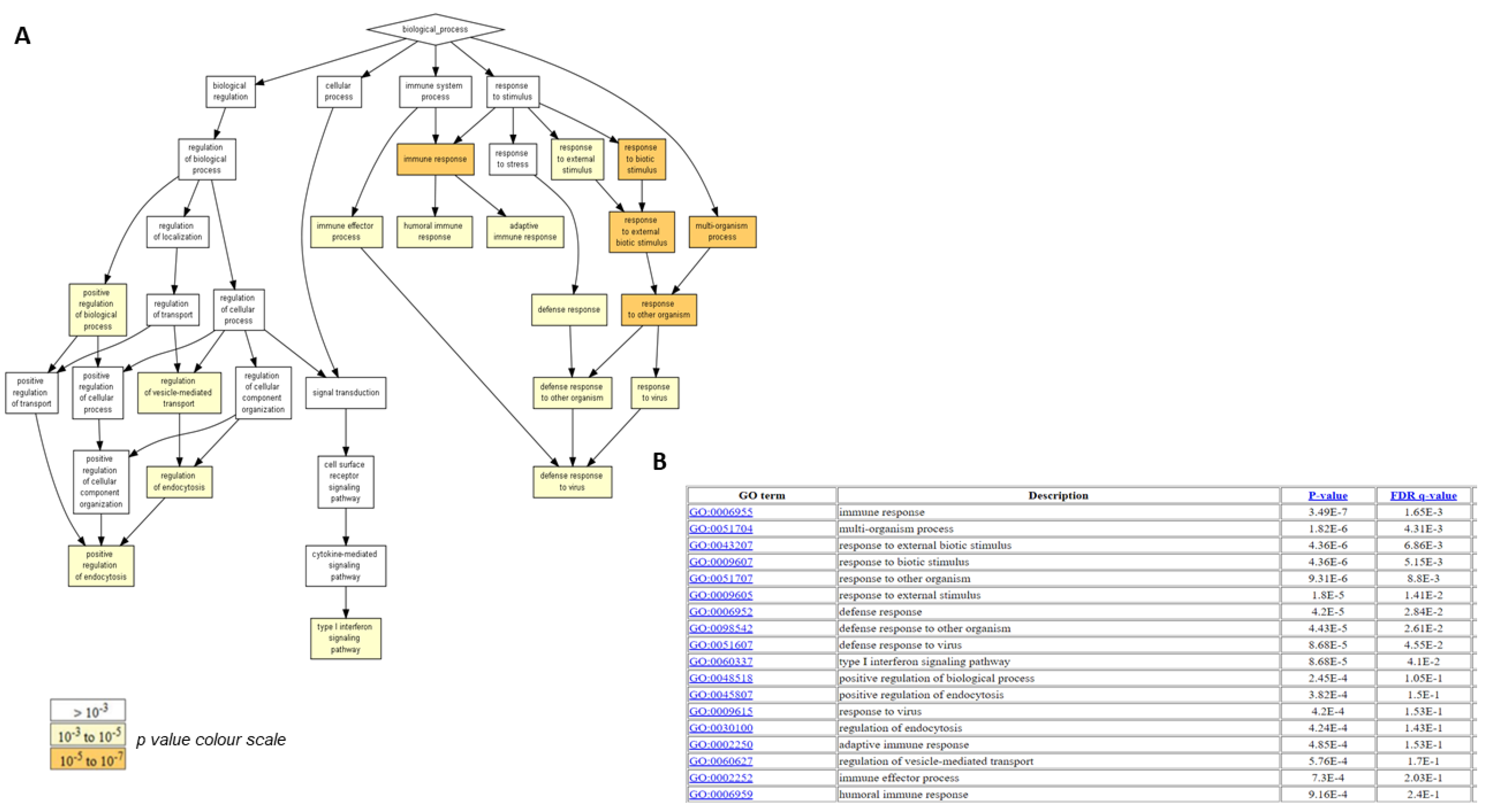
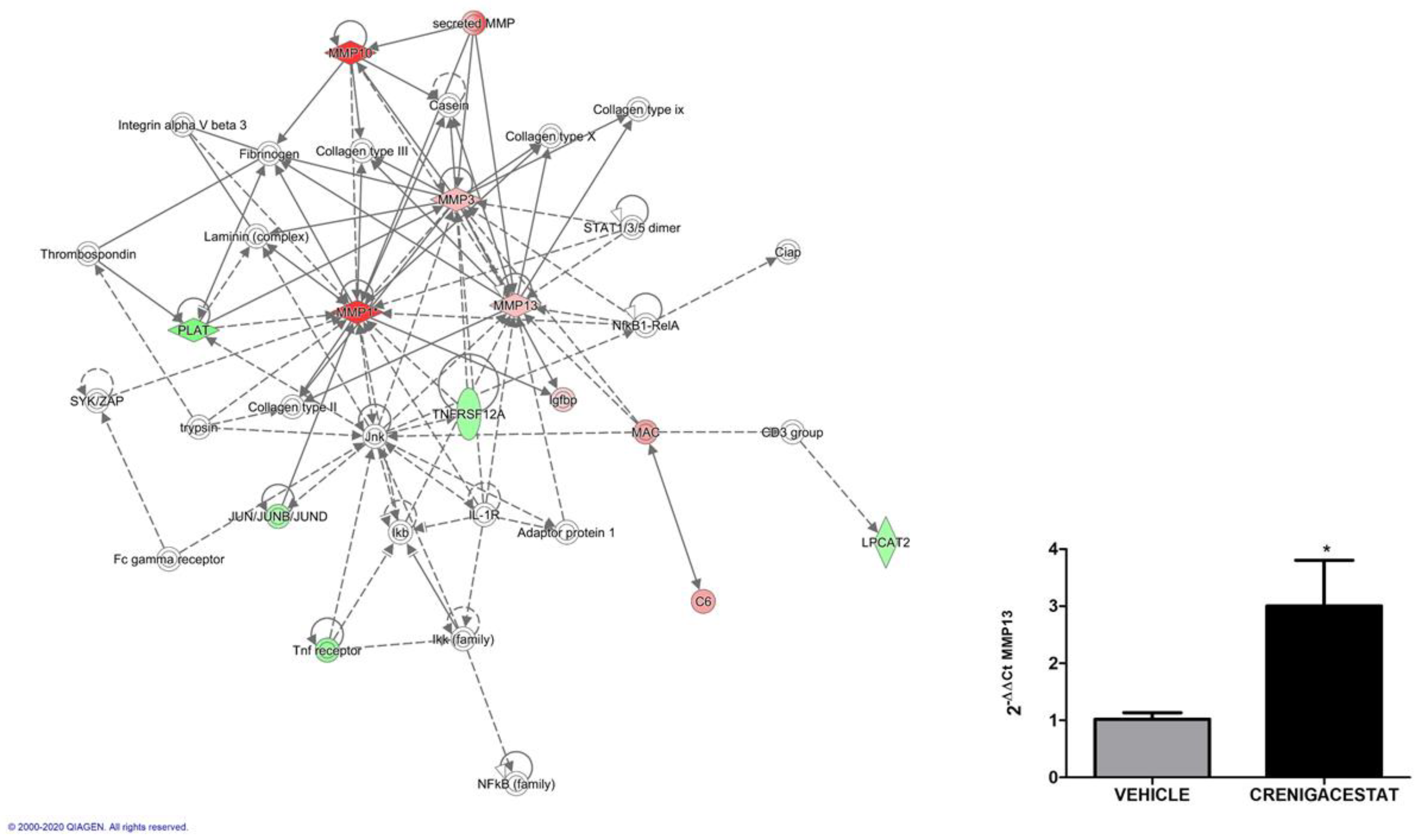
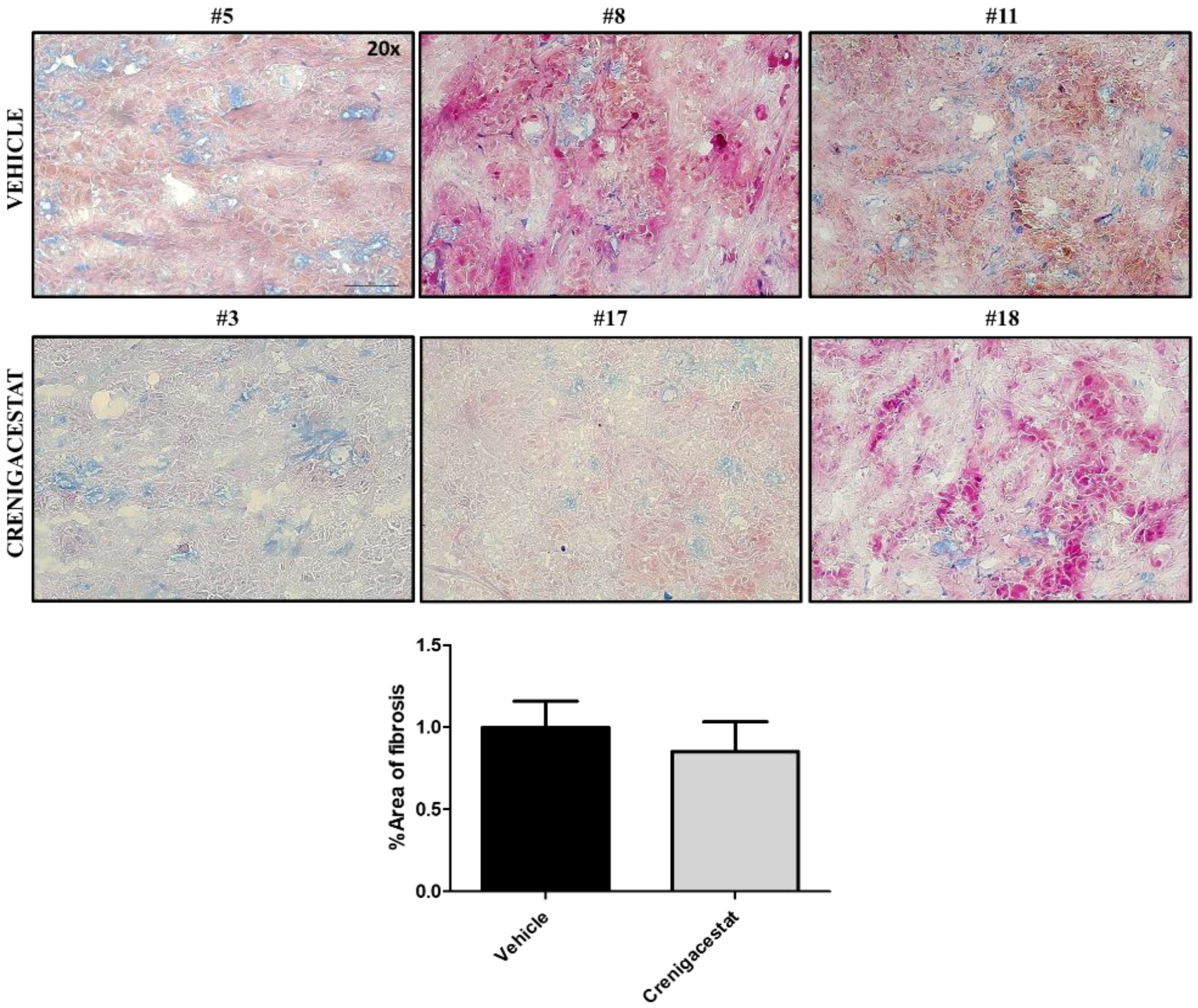
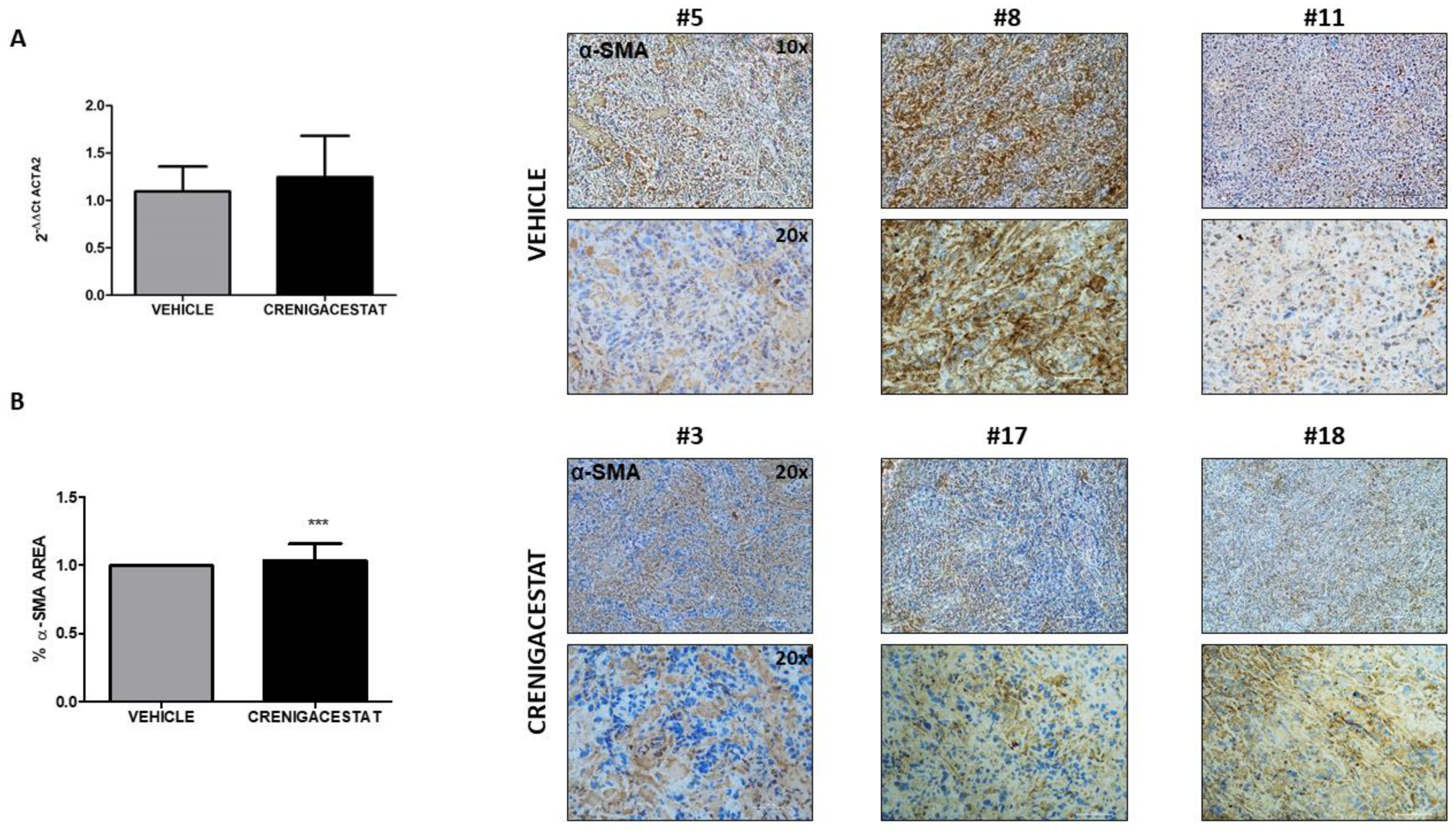
2.4. Biological Functions Regulated by Crenigacestat Treatment in the HuCCT1-Xenograft Mice Model
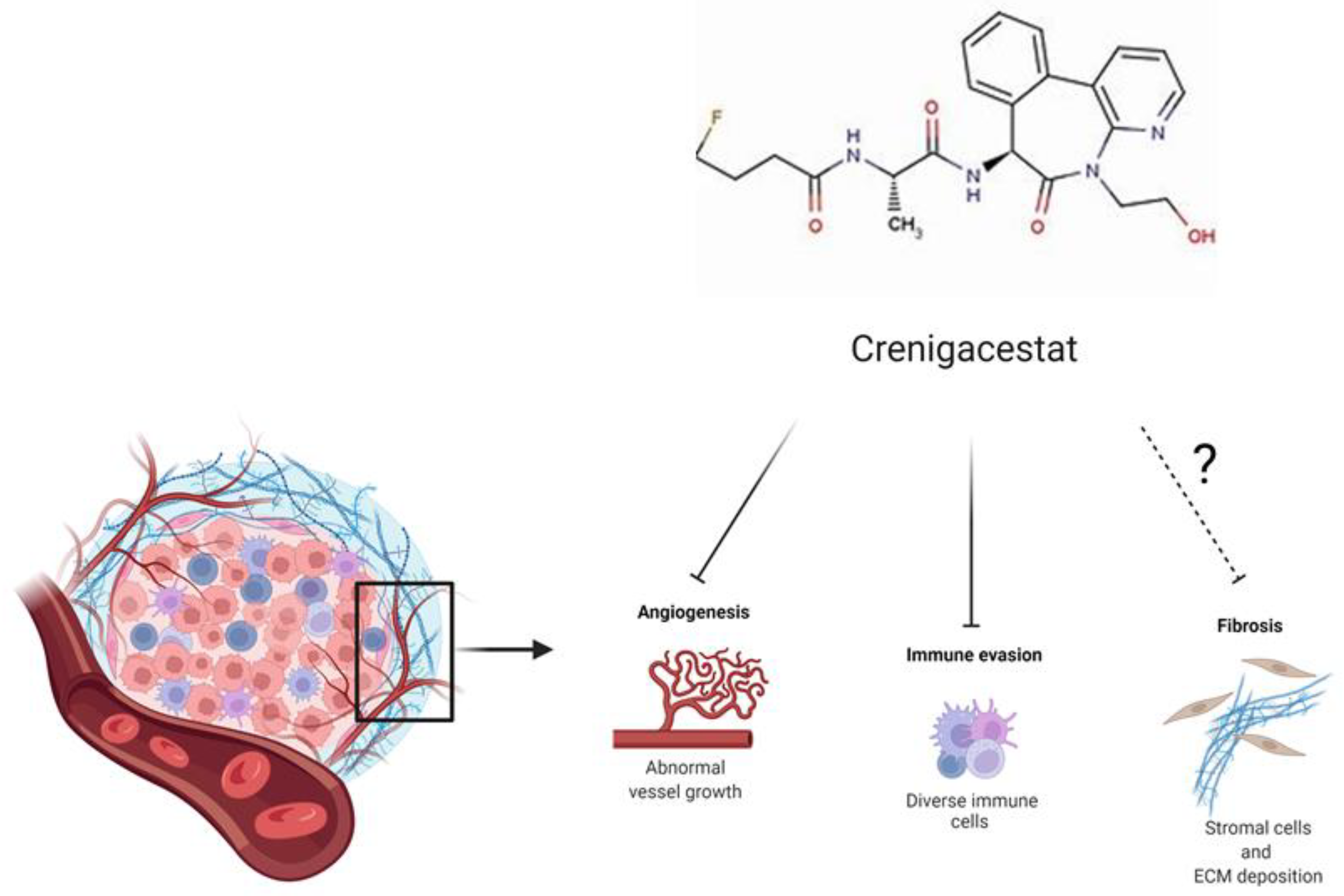
3. Discussion
4. Materials and Methods
4.1. In Vivo Study
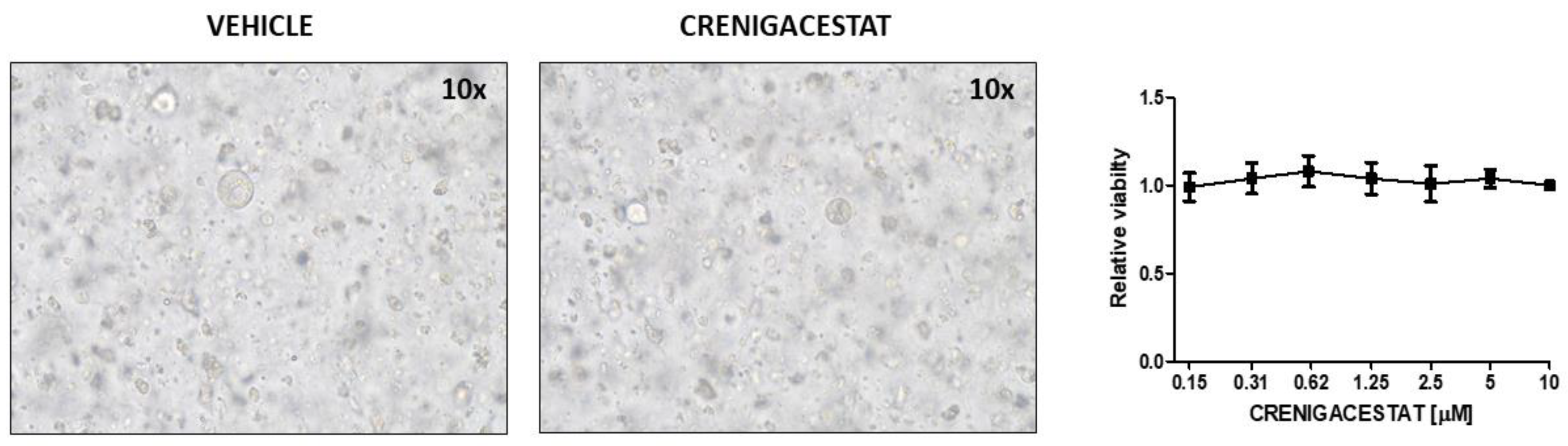
4.2. Establishment, Culture, and Treatment of Human iCCA-PDOs
4.3. Human iCCA-PDOs Treatment
4.4. Protein Extraction and Western Blot Analysis
4.5. RNA Extraction and cDNA Synthesis
4.6. Gene Expression Analysis
4.7. Quantitative Real-Time PCR
4.8. Histological Staining and Scoring
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAFs | Cancer-associated fibroblasts |
| DLL4 | Delta-like canonical Notch ligand 4 |
| ECM | Extracellular matrix |
| GO | Gene ontology |
| GSI | Gamma secretase inhibitor |
| HCC | Hepatocellular carcinoma |
| HES1 | Hairy/enhancer of split 1 |
| iCCA | Intrahepatic cholangiocarcinoma |
| IPA | Ingenuity pathway analysis |
| NICD | Notch intracellular domain |
| PDX | Patient-derived xenograft |
| VEGFA | Vascular endothelial growth factor A |
References
- Kirstein, M.M.; Vogel, A. Epidemiology and risk factors of cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Bertuccio, P.; Bosetti, C.; Levi, F.; Decarli, A.; Negri, E.; La Vecchia, C. A comparison of trends in mortality from primary liver cancer and intrahepatic cholangiocarcinoma in Europe. Ann. Oncol. 2013, 24, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, A.; Kim, J.U.; Eliahoo, J.; Taylor-Robinson, S.D.; Khan, S.A. Ablative therapy for unresectable intrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Clin. Exp. Hepatol. 2019, 9, 740–748. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The role of stroma in cholangiocarcinoma: The intriguing interplay between fibroblastic component, immune cell subsets and tumor epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Fabris, L.; Björkström, K.; Perugorria, M.J.; Cramer, T.; Mertens, J.; Lleo, A.; Sänger, H.; Lukacs, V.; Siebenhüner, A.; Moncsek, A.; et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019, 39, 63–78. [Google Scholar] [CrossRef]
- Loeuillard, E.; Conboy, C.B.; Gores, G.J.; Rizvi, S. Immunobiology of cholangiocarcinoma. JHEP Rep. 2019, 1, 297–311. [Google Scholar] [CrossRef]
- Baglieri, J.; Brenner, D.; Kisseleva, T. The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef]
- Fouassier, L.; Marzioni, M.; Afonso, M.B.; Dooley, S.; Gaston, K.; Giannelli, G.; Rodrigues, C.M.P.; Lozano, E.; Mancarella, S.; Segatto, O.; et al. Signalling networks in cholangiocarcinoma: Molecular pathogenesis, targeted therapies and drug resistance. Liver Int. 2019, 39, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. OncoTargets. Ther. 2013, 6, 1249. [Google Scholar] [PubMed]
- Gil-García, B.; Baladrón, V. The complex role of NOTCH receptors and their ligands in the development of hepatoblastoma, cholangiocarcinoma and hepatocellular carcinoma. Biol. Cell 2016, 108, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Geisler, F.; Strazzabosco, M. Emerging roles of Notch signaling in liver disease. Hepatology 2015, 61, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Precision medicine for human cancers with Notch signaling dysregulation (review). Int. J. Mol. Med. 2019, 45, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, Y.; Peng, B.; Liang, L.; Li, J. The roles of Notch1 expression in the migration of intrahepatic cholangiocarcinoma. BMC Cancer 2013, 13, 244. [Google Scholar] [CrossRef]
- Guo, J.; Fu, W.; Xiang, M.; Zhang, Y.; Zhou, K.; Xu, C.; Li, L.; Kuang, D.; Ye, F. Notch1 drives the formation and proliferation of intrahepatic cholangiocarcinoma. Curr. Med. Sci. 2019, 39, 929–937. [Google Scholar] [CrossRef]
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G. Crenigacestat, a selective NOTCH1 inhibitor, reduces intrahepatic cholangiocarcinoma progression by blocking VEGFA/DLL4/MMP13 axis. Cell Death Differ. 2020, 27, 2330–2343. [Google Scholar] [CrossRef]
- Mancarella, S.; Serino, G.; Gigante, I.; Cigliano, A.; Ribback, S.; Sanese, P.; Grossi, V.; Simone, C.; Armentano, R.; Evert, M.; et al. CD90 is regulated by notch1 and hallmarks a more aggressive intrahepatic cholangiocarcinoma phenotype. J. Exp. Clin. Cancer Res. 2022, 41, 65. [Google Scholar] [CrossRef]
- Cigliano, A.; Wang, J.; Chen, X.; Calvisi, D.F. Role of the Notch signaling in cholangiocarcinoma. Expert Opin. Ther. Targets 2017, 21, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E. Immunodeficient mouse models: An overview. Open Immunol. J. 2009, 2, 79–85. [Google Scholar] [CrossRef]
- Sulpice, L.; Desille, M.; Turlin, B.; Fautrel, A.; Boudjema, K.; Clément, B.; Coulouarn, C. Gene expression profiling of the tumor microenvironment in human intrahepatic cholangiocarcinoma. Genom. Data 2016, 7, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, L.; Rayar, M.; Desille, M.; Turlin, B.; Fautrel, A.; Boucher, E.; Llamas-Gutierrez, F.; Meunier, B.; Boudjema, K.; Clément, B.; et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2013, 58, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Fingleton, B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 2036–2042. [Google Scholar] [CrossRef]
- MANICONE, A.; MCGUIRE, J. Matrix metalloproteinases as modulators of inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar] [CrossRef]
- Yamazaki, K.; Masugi, Y.; Sakamoto, M. Molecular pathogenesis of hepatocellular carcinoma: Altering transforming growth factor-β signaling in hepatocarcinogenesis. Dig. Dis. 2011, 29, 284–288. [Google Scholar] [CrossRef]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Dituri, F.; Giannelli, G. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/α3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology 2016, 64, 2103–2117. [Google Scholar] [CrossRef]
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G.; Agarwal, R.; et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cancers 2019, 39, 43–62. [Google Scholar]
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Intrahepatic Cholangiocarcinoma: Continuing Challenges and Translational Advances. Hepatology 2019, 69, 1803–1815. [Google Scholar]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metallo1. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated fibroblast program orchestrates tumor initiation and progression; Molecular mechanisms and the associated therapeutic strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, P. Tumor microenvironment and angiogenesis. Front. Biosci. 2008, 13, 6537–6553. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the tissue microenvironment: Relevance in fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as multifaceted orchestrator in HCC progression: Signaling, EMT, immune microenvironment, and novel therapeutic perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar]
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef]
- Mancarella, S.; Krol, S.; Crovace, A.; Leporatti, S.; Dituri, F.; Frusciante, M.; Giannelli, G. Validation of hepatocellular carcinoma experimental models for TGF-β promoting tumor progression. Cancers 2019, 11, 1510. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Emig, D.; Salomonis, N.; Baumbach, J.; Lengauer, T.; Conklin, B.R.; Albrecht, M. AltAnalyze and DomainGraph: Analyzing and visualizing exon expression data. Nucleic Acids Res. 2010, 38, W755–W762. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Top Diseases and Functions | Score | Focus Molecules |
|---|---|---|---|
| 1 | Cancer, gastrointestinal disease, organism injury and abnormalities | 55 | 31 |
| 2 | Cancer, cell morphology, tissue development | 38 | 24 |
| 3 | Cell morphology, embryonic development, hair and skin development and function | 33 | 22 |
| 4 | Metabolic disease, protein degradation, protein synthesis | 33 | 22 |
| 5 | Carbohydrate metabolism, cardiovascular disease, post-translational modification | 31 | 21 |
| 6 | Cell death and survival, lipid metabolism, nervous system development and function | 31 | 21 |
| 7 | Cellular development, cellular growth and proliferation, cellular movement | 29 | 20 |
| 8 | Dermatological diseases and conditions, immunological disease, organismal injury and abnormalities | 25 | 18 |
| 9 | Developmental disorder, organism injury and abnormalities, renal and urological disease | 23 | 17 |
| 10 | Cancer, cell cycle, gene expression | 23 | 17 |
| 11 | Embryonic development, hematological system development and function, lymphoid tissue structure and development | 21 | 16 |
| 12 | Cancer, dermatological diseases and conditions, organism injury and abnormalities | 21 | 16 |
| 13 | Cellular assembly and organization, lipid metabolism, small molecule biochemistry | 21 | 16 |
| 14 | Auditory disease, hereditary disorder, neurological disease | 21 | 16 |
| 15 | Dermatological diseases and conditions, immunological disease, inflammatory disease | 19 | 15 |
| 16 | Cell morphology, embryonic development, nervous system development and function | 19 | 15 |
| 17 | Cancer, cellular assembly and organization, connective tissue disorders | 18 | 14 |
| 18 | Cell morphology, organ morphology, organism injury and abnormalities | 16 | 13 |
| 19 | Drug metabolism, increased levels of AST, lipid metabolism | 14 | 12 |
| 20 | Cardiovascular disease, cellular compromise, organism injury and abnormalities | 14 | 12 |
| 21 | Embryonic development, organism development, tissue morphology | 13 | 11 |
| 22 | Cell cycle, cellular movement, infectious diseases | 13 | 11 |
| 23 | Connective tissue development and function, connective tissue disorders, organism injury and abnormalities | 8 | 8 |
| 24 | Cellular movement, immune cell trafficking, inflammatory response | 7 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancarella, S.; Serino, G.; Coletta, S.; Armentano, R.; Dituri, F.; Ardito, F.; Ruzzenente, A.; Fabregat, I.; Giannelli, G. The Tumor Microenvironment Drives Intrahepatic Cholangiocarcinoma Progression. Int. J. Mol. Sci. 2022, 23, 4187. https://doi.org/10.3390/ijms23084187
Mancarella S, Serino G, Coletta S, Armentano R, Dituri F, Ardito F, Ruzzenente A, Fabregat I, Giannelli G. The Tumor Microenvironment Drives Intrahepatic Cholangiocarcinoma Progression. International Journal of Molecular Sciences. 2022; 23(8):4187. https://doi.org/10.3390/ijms23084187
Chicago/Turabian StyleMancarella, Serena, Grazia Serino, Sergio Coletta, Raffaele Armentano, Francesco Dituri, Francesco Ardito, Andrea Ruzzenente, Isabel Fabregat, and Gianluigi Giannelli. 2022. "The Tumor Microenvironment Drives Intrahepatic Cholangiocarcinoma Progression" International Journal of Molecular Sciences 23, no. 8: 4187. https://doi.org/10.3390/ijms23084187
APA StyleMancarella, S., Serino, G., Coletta, S., Armentano, R., Dituri, F., Ardito, F., Ruzzenente, A., Fabregat, I., & Giannelli, G. (2022). The Tumor Microenvironment Drives Intrahepatic Cholangiocarcinoma Progression. International Journal of Molecular Sciences, 23(8), 4187. https://doi.org/10.3390/ijms23084187