
The Morpho-Molecular Landscape of Spitz Neoplasms



Abstract
:1. Introduction
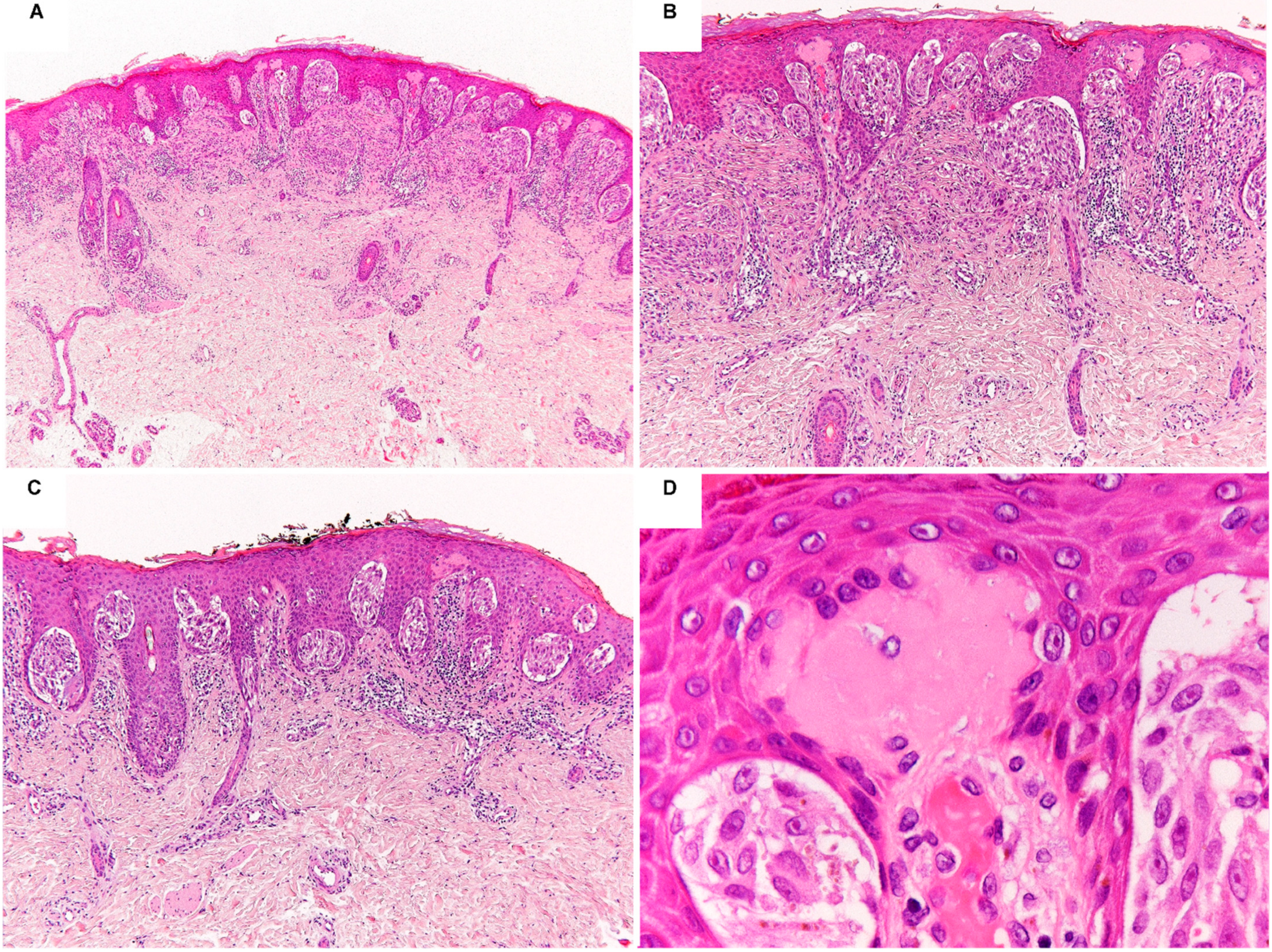
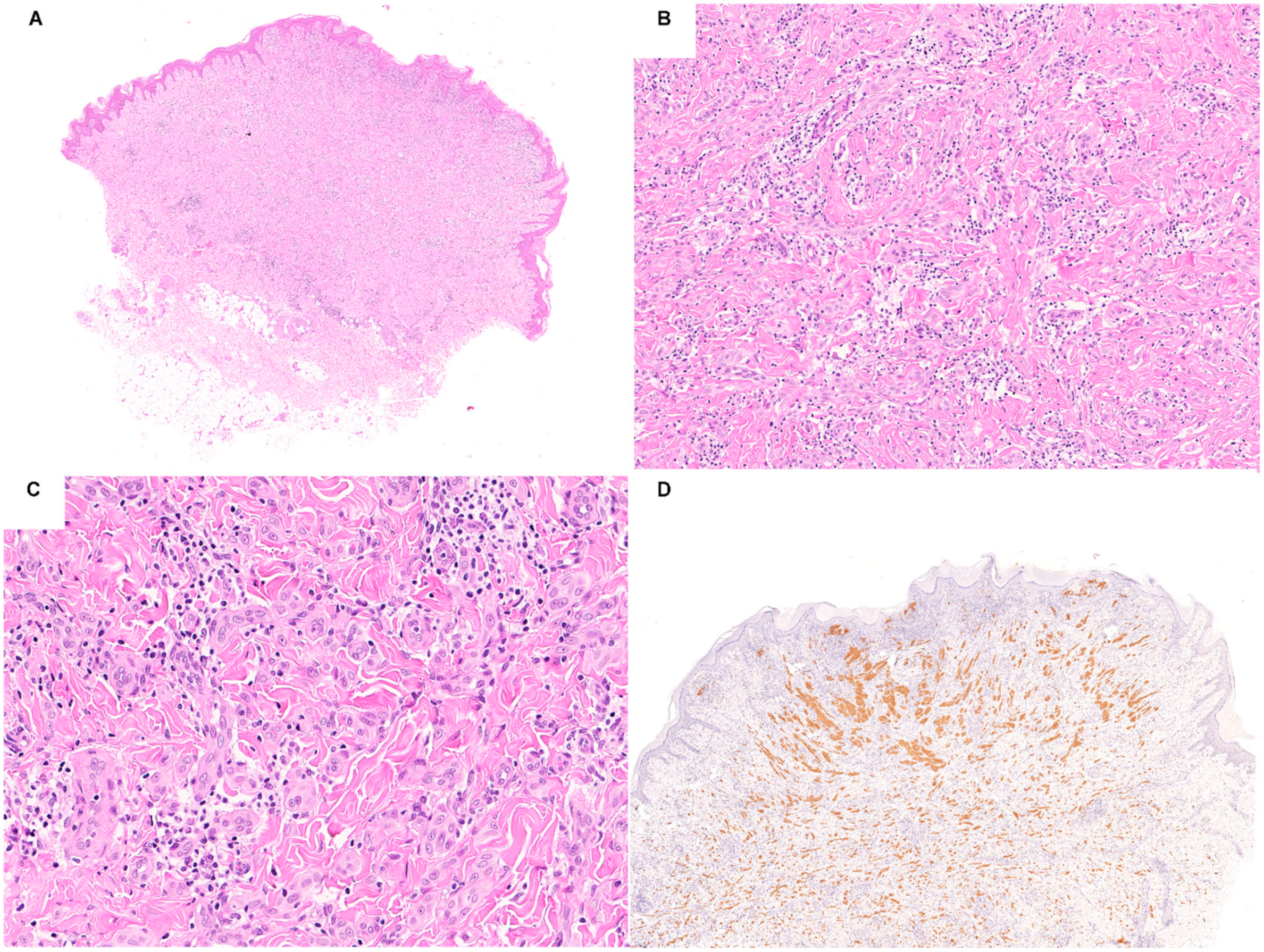
2. Spitz Neoplasms with HRAS Mutations or 11p Copy Number Gains
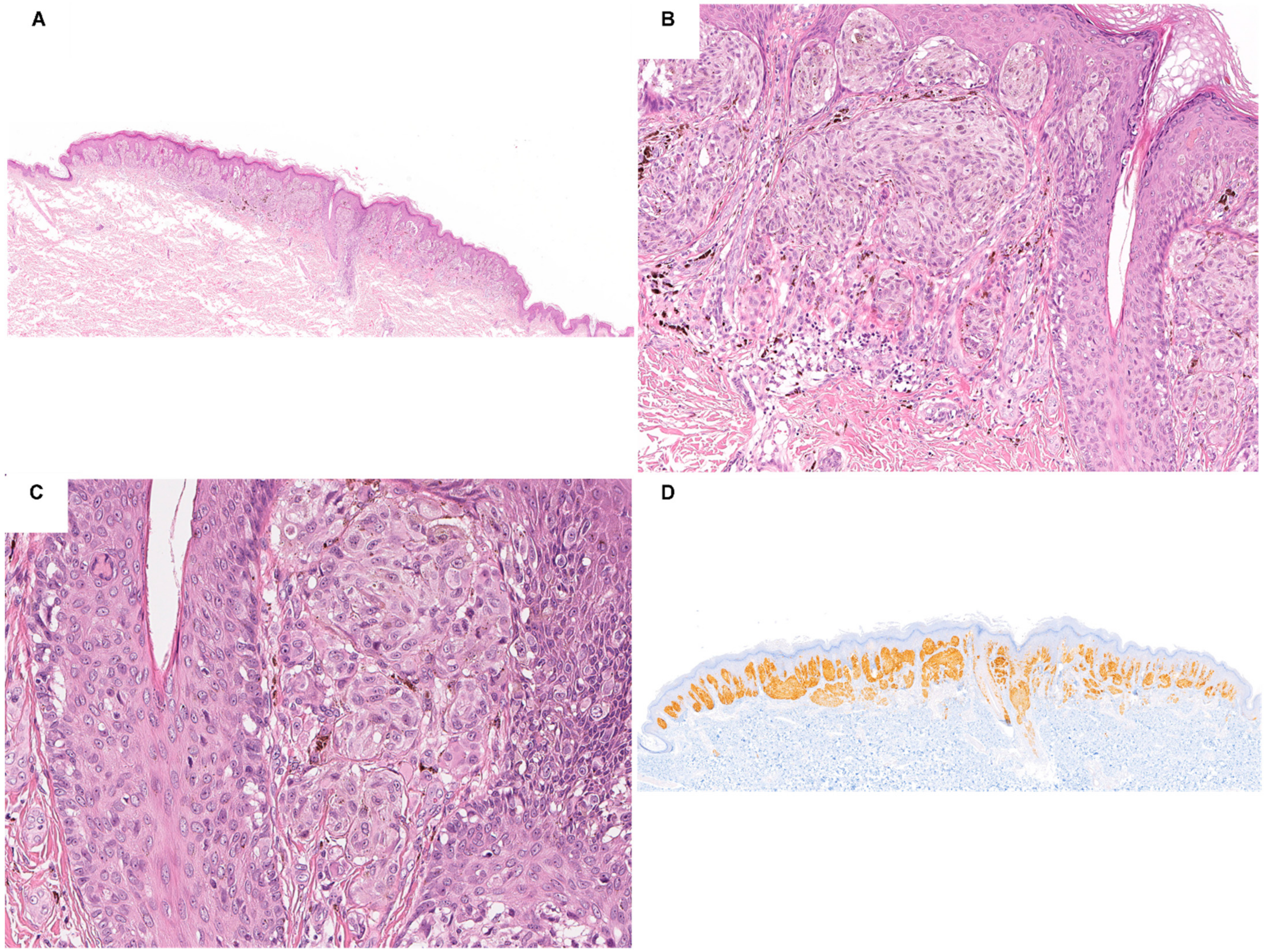
3. Spitz Neoplasms with ALK Fusions
4. Spitz Neoplasms with ROS1 Fusions
5. Spitz Neoplasms with NTRK Fusions
5.1. Spitz Neoplasms with NTRK1 Fusions
5.2. Spitz Neoplasms with NTRK2 Fusions
5.3. Spitz Neoplasms with NTRK3 Fusions
6. Spitz Neoplasms with RET Fusions
7. Spitz Neoplasms with MET Fusions
8. Spitz Neoplasms with MAP2K1 Mutations
9. Spitz Neoplasms with MAP3K8 Fusions
10. Spitz Neoplasms with BRAF Fusions
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. 2014, 9, 239–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitz, S. Melanomas of childhood. Am. J. Pathol. 1948, 24, 591–609. [Google Scholar] [PubMed]
- Quan, V.L.; Panah, E.; Zhang, B.; Shi, K.; Mohan, L.S.; Gerami, P. The role of gene fusions in melanocytic neoplasms. J. Cutan. Pathol. 2019, 46, 878–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, R.C.; Chalet, M.D.; Ackerman, A.B. Benign juvenile melanoma (Spitz nevus) vs. superficial spreading malignant melanoma: Criteria for histologic differentiation. J. Dermatol. Surg. 1975, 1, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.S.; Peternel, S.; Mully, T.W.; North, J.P.; Pincus, L.B.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; Yeh, I. Spitz melanoma is a distinct subset of spitzoid melanoma. Mod. Pathol. 2020, 33, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Sulzberger, M.B.; Kopf, A.W.; Witten, V.H. Pigmented nevi, benign juvenile melanoma and circumscribed precancerous melanosis. Postgrad. Med. 1959, 26, 617–631. [Google Scholar] [CrossRef]
- Kernen, J.A.; Ackerman, L.V. Spindle cell nevi and epithelioid cell nevi (so-called juvenile melanomas) in children and adults: A clinicopathological study of 27 cases. Cancer 1960, 13, 612–625. [Google Scholar] [CrossRef]
- Requena, C.; Requena, L.; Kutzner, H.; Sánchez Yus, E. Spitz nevus: A clinicopathological study of 349 cases. Am. J. Dermatopathol. 2009, 31, 107–116. [Google Scholar] [CrossRef]
- Weedon, D.; Little, J.H. Spindle and epithelioid cell nevi in children and adults. A review of 211 cases of the Spitz nevus. Cancer 1977, 40, 217–225. [Google Scholar] [CrossRef]
- Massi, D.; De Giorgi, V.; Mandalà, M. The complex management of atypical Spitz tumours. Pathology 2016, 48, 132–141. [Google Scholar] [CrossRef]
- Amin, S.M.; Haugh, A.M.; Lee, C.Y.; Zhang, B.; Bubley, J.A.; Merkel, E.A.; Verzì, A.E.; Gerami, P. A Comparison of Morphologic and Molecular Features of BRAF, ALK, and NTRK1 Fusion Spitzoid Neoplasms. Am. J. Surg. Pathol. 2017, 41, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Bastian, B.C.; LeBoit, P.E.; Pinkel, D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am. J. Pathol. 2000, 157, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, D.E.; Massi, D.; Scolyer, R.A.; Willemze, R.; World Health Organization. WHO Classification of Skin Tumours, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2018; p. 470. [Google Scholar]
- Urso, C. Melanocytic Skin Neoplasms: What Lesson from Genomic Aberrations? Am. J. Dermatopathol. 2019, 41, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Potrony, M.; Badenas, C.; Aguilera, P.; Puig-Butille, J.A.; Carrera, C.; Malvehy, J.; Puig, S. Update in genetic susceptibility in melanoma. Ann. Transl. Med. 2015, 3, 210. [Google Scholar] [CrossRef]
- Gill, M.; Renwick, N.; Silvers, D.N.; Celebi, J.T. Lack of BRAF mutations in Spitz nevi. J. Investig. Dermatol. 2004, 122, 1325–1326. [Google Scholar] [CrossRef] [Green Version]
- Palmedo, G.; Hantschke, M.; Rütten, A.; Mentzel, T.; Hügel, H.; Flaig, M.J.; Yazdi, A.S.; Sander, C.A.; Kutzner, H. The T1796A mutation of the BRAF gene is absent in Spitz nevi. J. Cutan. Pathol. 2004, 31, 266–270. [Google Scholar] [CrossRef]
- Van Dijk, M.C.; Bernsen, M.R.; Ruiter, D.J. Analysis of mutations in B-RAF, N-RAS, and H-RAS genes in the differential diagnosis of Spitz nevus and spitzoid melanoma. Am. J. Surg. Pathol. 2005, 29, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Curtin, J.A.; Pinkel, D.; Bastian, B.C. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J. Investig. Dermatol. 2007, 127, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Botton, T.; Yeh, I.; Nelson, T.; Vemula, S.S.; Sparatta, A.; Garrido, M.C.; Allegra, M.; Rocchi, S.; Bahadoran, P.; McCalmont, T.H.; et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment. Cell Melanoma Res. 2013, 26, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; Botton, T.; Talevich, E.; Shain, A.H.; Sparatta, A.J.; de la Fouchardiere, A.; Mully, T.W.; North, J.P.; Garrido, M.C.; Gagnon, A.; et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat. Commun. 2015, 6, 7174. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; Tee, M.K.; Botton, T.; Shain, A.H.; Sparatta, A.J.; Gagnon, A.; Vemula, S.S.; Garrido, M.C.; Nakamaru, K.; Isoyama, T.; et al. NTRK3 kinase fusions in Spitz tumours. J. Pathol. 2016, 240, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Yeh, I.; Busam, K.J.; McCalmont, T.H.; LeBoit, P.E.; Pissaloux, D.; Alberti, L.; de la Fouchardière, A.; Bastian, B.C. Filigree-like Rete Ridges, Lobulated Nests, Rosette-like Structures, and Exaggerated Maturation Characterize Spitz Tumors with NTRK1 Fusion. Am. J. Surg. Pathol. 2019, 43, 737–746. [Google Scholar] [CrossRef]
- de la Fouchardière, A.; Tee, M.K.; Peternel, S.; Valdebran, M.; Pissaloux, D.; Tirode, F.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C.; et al. Fusion partners of NTRK3 affect subcellular localization of the fusion kinase and cytomorphology of melanocytes. Mod. Pathol. 2021, 34, 735–747. [Google Scholar] [CrossRef]
- Goto, K.; Pissaloux, D.; Tirode, F.; de la Fouchardière, A. Spitz nevus with a novel TFG-NTRK2 fusion: The first case report of NTRK2-rearranged Spitz/Reed nevus. J. Cutan. Pathol. 2021, 48, 1193–1196. [Google Scholar] [CrossRef]
- Quan, V.L.; Zhang, B.; Zhang, Y.; Mohan, L.S.; Shi, K.; Wagner, A.; Kruse, L.; Taxter, T.; Beaubier, N.; White, K.; et al. Integrating Next-Generation Sequencing with Morphology Improves Prognostic and Biologic Classification of Spitz Neoplasms. J. Investig. Dermatol. 2020, 140, 1599–1608. [Google Scholar] [CrossRef]
- Houlier, A.; Pissaloux, D.; Masse, I.; Tirode, F.; Karanian, M.; Pincus, L.B.; McCalmont, T.H.; LeBoit, P.E.; Bastian, B.C.; Yeh, I.; et al. Melanocytic tumors with MAP3K8 fusions: Report of 33 cases with morphological-genetic correlations. Mod. Pathol. 2020, 33, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Kervarrec, T.; Pissaloux, D.; Tirode, F.; Samimi, M.; Jacquemus, J.; Castillo, C.; de la Fouchardière, A. Morphologic features in a series of 352 Spitz melanocytic proliferations help predict their oncogenic drivers. Virchows Arch. 2021, 480, 369–382. [Google Scholar] [CrossRef]
- Quan, V.L.; Zhang, B.; Mohan, L.S.; Shi, K.; Isales, M.C.; Panah, E.; Taxter, T.J.; Beaubier, N.; White, K.; Gerami, P. Activating Structural Alterations in MAPK Genes Are Distinct Genetic Drivers in a Unique Subgroup of Spitzoid Neoplasms. Am. J. Surg. Pathol. 2019, 43, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Kerckhoffs, K.G.P.; Aallali, T.; Ambarus, C.A.; Sigurdsson, V.; Jansen, A.M.L.; Blokx, W.A.M. Expanding spectrum of “spitzoid” lesions: A small series of 4 cases with MAP2K1 mutations. Virchows Arch. 2021, 479, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Kim, D.; Zhang, B.; Compres, E.V.; Khan, A.U.; Busam, K.J.; Gerami, P. Melanocytic Neoplasms with MAP2K1 in Frame Deletions and Spitz Morphology. Am. J. Dermatopathol. 2020, 42, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.; Nosek, D.; Waldenbäck, P.; Martinek, P.; Jonsson, B.A.; Galgonkova, P.; Hawawrehova, M.; Berouskova, P.; Kastnerova, L.; Persichetti, P.; et al. MAP2K1-Mutated Melanocytic Neoplasms With a SPARK-Like Morphology. Am. J. Dermatopathol. 2021, 43, 412–417. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Maffeis, V.; Nicolè, L.; Cappellesso, R. RAS, Cellular Plasticity, and Tumor Budding in Colorectal Cancer. Front. Oncol. 2019, 9, 1255. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Roy, S.; Apolloni, A.; Lane, A.; Hancock, J.F. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 1998, 273, 24052–24056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, A.L.; Sanchez, M.I.; Grichnik, J.M. Molecular nevogenesis. Dermatol. Res. Pract. 2011, 2011, 463184. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Kutzner, H.; Cerroni, L.; Mihm, M.C.; Busam, K.J.; Murali, R. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology 2016, 48, 113–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastian, B.C.; LeBoit, P.E.; Hamm, H.; Bröcker, E.B.; Pinkel, D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998, 58, 2170–2175. [Google Scholar] [PubMed]
- Bastian, B.C.; Wesselmann, U.; Pinkel, D.; Leboit, P.E. Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J. Investig. Dermatol. 1999, 113, 1065–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, L.; Helm, T.; Cheney, R.; Conroy, J.; Sait, S.; Guitart, J.; Gerami, P. Correlating array comparative genomic hybridization findings with histology and outcome in spitzoid melanocytic neoplasms. Int. J. Clin. Exp. Pathol. 2010, 3, 593–599. [Google Scholar]
- Van Engen-Van Grunsven, A.C.; van Dijk, M.C.; Ruiter, D.J.; Klaasen, A.; Mooi, W.J.; Blokx, W.A. HRAS-mutated Spitz tumors: A subtype of Spitz tumors with distinct features. Am. J. Surg. Pathol. 2010, 34, 1436–1441. [Google Scholar] [CrossRef]
- Lazova, R.; Pornputtapong, N.; Halaban, R.; Bosenberg, M.; Bai, Y.; Chai, H.; Krauthammer, M. Spitz nevi and Spitzoid melanomas: Exome sequencing and comparison with conventional melanocytic nevi and melanomas. Mod. Pathol. 2017, 30, 640–649. [Google Scholar] [CrossRef] [Green Version]
- Bender, R.P.; McGinniss, M.J.; Esmay, P.; Velazquez, E.F.; Reimann, J.D. Identification of HRAS mutations and absence of GNAQ or GNA11 mutations in deep penetrating nevi. Mod. Pathol. 2013, 26, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Kiyohara, T.; Sawai, T.; Kumakiri, M. Proliferative nodule in small congenital melanocytic naevus after childhood. Acta Derm. Venereol. 2012, 92, 96–97. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Berger, M.F.; Marghoob, A.; Bhanot, U.K.; Toyohara, J.P.; Pulitzer, M.P. Combined melanocytic and sweat gland neoplasm: Cell subsets harbor an identical HRAS mutation in phacomatosis pigmentokeratotica. J. Cutan. Pathol. 2014, 41, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, K.; Szabo, S.; Cottrell, C.E.; McNulty, S.M.; Segura, A.; Sokumbi, O.; Browning, M.; Siegel, D.H. Mosaic pathogenic HRAS variant in a patient with nevus spilus with agminated Spitz nevi and parametrial-uterine rhabdomyosarcoma. Br. J. Dermatol. 2018, 178, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Pontoizeau, J.; Stefan, A.; Comoz, F.; Houlier, A.; Haddad, V.; Pissaloux, D.; de la Fouchardiere, A. Agminated Spitz nevus arising in normal skin with redundant HRAS mutation. Eur. J. Dermatol. 2017, 27, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, C.; Teer, J.K.; Zhang, Y.; Deschaine, M.; Sondak, V.K.; Messina, J.L. Genomic analysis of a case of agminated Spitz nevi and congenital-pattern nevi arising in extensive nevus spilus. J. Cutan. Pathol. 2018, 45, 180–183. [Google Scholar] [CrossRef]
- Sarin, K.Y.; Sun, B.K.; Bangs, C.D.; Cherry, A.; Swetter, S.M.; Kim, J.; Khavari, P.A. Activating HRAS mutation in agminated Spitz nevi arising in a nevus spilus. JAMA Dermatol. 2013, 149, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Massi, D.; Simi, L.; Sensi, E.; Baroni, G.; Xue, G.; Scatena, C.; Caldarella, A.; Pinzani, P.; Fontanini, G.; Carobbio, A.; et al. Immunohistochemistry is highly sensitive and specific for the detection of NRASQ61R mutation in melanoma. Mod. Pathol. 2015, 28, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Felisiak-Goląbek, A.; Inaguma, S.; Kowalik, A.; Wasąg, B.; Wang, Z.F.; Zięba, S.; Pięciak, L.; Ryś, J.; Kopczynski, J.; Sarlomo-Rikala, M.; et al. SP174 Antibody Lacks Specificity for NRAS Q61R and Cross-Reacts with HRAS and KRAS Q61R Mutant Proteins in Malignant Melanoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 40–45. [Google Scholar] [CrossRef]
- Gerami, P.; Kim, D.; Compres, E.V.; Zhang, B.; Khan, A.U.; Sunshine, J.C.; Quan, V.L.; Busam, K. Clinical, morphologic, and genomic findings in ROS1 fusion Spitz neoplasms. Mod. Pathol. 2021, 34, 348–357. [Google Scholar] [CrossRef]
- Da Forno, P.D.; Pringle, J.H.; Fletcher, A.; Bamford, M.; Su, L.; Potter, L.; Saldanha, G. BRAF, NRAS and HRAS mutations in spitzoid tumours and their possible pathogenetic significance. Br. J. Dermatol. 2009, 161, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Hillen, L.M.; Van den Oord, J.; Geybels, M.S.; Becker, J.C.; Zur Hausen, A.; Winnepenninckx, V. Genomic Landscape of Spitzoid Neoplasms Impacting Patient Management. Front. Med. 2018, 5, 344. [Google Scholar] [CrossRef]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulford, K.; Morris, S.W.; Turturro, F. Anaplastic lymphoma kinase proteins in growth control and cancer. J. Cell Physiol. 2004, 199, 330–358. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Nieborowska-Skorska, M.; Hoser, G.; Morrione, A.; Majewski, M.; Xue, L.; Morris, S.W.; Wasik, M.A.; Skorski, T. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001, 61, 2194–2199. [Google Scholar] [PubMed]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Saraggi, D.; Salmaso, R.; Zamuner, C.; Munari, G.; Lanza, C.; Alaibac, M.S.; Bassetto, F.; Rugge, M.; Montesco, M.C.; Cerroni, L.; et al. Prevalence of ALK gene alterations among the spectrum of plexiform spitzoid lesions. J. Am. Acad. Dermatol. 2018, 79, 728–735. [Google Scholar] [CrossRef]
- Abounader, R.; Reznik, T.; Colantuoni, C.; Martinez-Murillo, F.; Rosen, E.M.; Laterra, J. Regulation of c-Met-dependent gene expression by PTEN. Oncogene 2004, 23, 9173–9182. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Barnhill, R.L.; Dummer, R.; Dalton, J.; Wu, J.; Pappo, A.; Bahrami, A. TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci. Rep. 2015, 5, 11200. [Google Scholar] [CrossRef] [Green Version]
- Kastnerova, L.; Martinek, P.; Grossmann, P.; Steiner, P.; Vanecek, T.; Kyclova, J.; Ferak, I.; Zalud, R.; Slehobr, O.; Svajdler, P.; et al. A Clinicopathological Study of 29 Spitzoid Melanocytic Lesions with ALK Fusions, Including Novel Fusion Variants, Accompanied by Fluorescence In Situ Hybridization Analysis for Chromosomal Copy Number Changes, and Both TERT Promoter and Next-Generation Sequencing Mutation Analysis. Am. J. Dermatopathol. 2020, 42, 578–592. [Google Scholar] [CrossRef]
- Yeh, I.; de la Fouchardiere, A.; Pissaloux, D.; Mully, T.W.; Garrido, M.C.; Vemula, S.S.; Busam, K.J.; LeBoit, P.E.; McCalmont, T.H.; Bastian, B.C. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2015, 39, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Busam, K.J.; Kutzner, H.; Cerroni, L.; Wiesner, T. Clinical and Pathologic findings of Spitz nevi and atypical Spitz tumors with ALK fusions. Am. J. Surg. Pathol. 2014, 38, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Melchers, R.C.; Willemze, R.; van Doorn, R.; Jansen, P.M.; Cleven, A.H.G.; Solleveld, N.; Vermeer, M.H.; Quint, K.D. Corresponding anaplastic lymphoma kinase-tropomyosin 3. JAAD Case Rep. 2019, 5, 970–972. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.; Fan, L.; Pribnow, A.; Silkov, A.; Rice, S.V.; Lee, S.; Shao, Y.; Shaner, B.; Mulder, H.; Nakitandwe, J.; et al. Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat. Med. 2019, 25, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Togashi, Y.; Matsuzaki, I.; Baba, S.; Takeuchi, K.; Inaba, Y.; Jinnin, M.; Murata, S.I. A case report of atypical Spitz tumor harboring a novel MLPH-ALK gene fusion with discordant ALK immunohistochemistry results. Hum. Pathol. 2018, 80, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.T.; Marrano, P.; Swanson, D.; Dickson, B.C.; Thorner, P.S. Fusion of ALK to the melanophilin gene MLPH in pediatric Spitz nevi. Hum. Pathol. 2019, 87, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Jungbluth, A.; Kutzner, H.; Wiesner, T.; Busam, K.J. Spitz Tumors: Comparison of Histological Features in Relationship to Immunohistochemical Staining for ALK and NTRK1. Int. J. Surg. Pathol. 2016, 24, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Wang, J.Y.; Raghavan, S.S.; Zhang, J.; Wan, D.C.; Born, D.; Koo, M.; Hazard, F.K.; Novoa, R.A.; Rieger, K.E. ALK-positive compound Spitz nevus with extensive perineural and intraneural neurotropism. J. Cutan. Pathol. 2021, 48, 154–159. [Google Scholar] [CrossRef]
- Drilon, A.; Jenkins, C.; Iyer, S.; Schoenfeld, A.; Keddy, C.; Davare, M.A. ROS1-dependent cancers-biology, diagnostics and therapeutics. Nat. Rev. Clin. Oncol. 2021, 18, 35–55. [Google Scholar] [CrossRef]
- Donati, M.; Kastnerova, L.; Martinek, P.; Grossmann, P.; Sticová, E.; Hadravský, L.; Torday, T.; Kyclova, J.; Michal, M.; Kazakov, D.V. Spitz Tumors With ROS1 Fusions: A ClinicoPathological Study of 6 Cases, Including FISH for Chromosomal Copy Number Alterations and Mutation Analysis Using Next-Generation Sequencing. Am. J. Dermatopathol. 2020, 42, 92–102. [Google Scholar] [CrossRef]
- Cesinaro, A.M.; Gallo, G.; Manfredini, S.; Maiorana, A.; Bettelli, S.R. ROS1 pattern of immunostaining in 11 cases of spitzoid tumour: Comparison with histoPathological, fluorescence in-situ hybridisation and next-generation sequencing analysis. Histopathology 2021, 79, 966–974. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Rubin, J.B.; Segal, R.A. Growth, survival and migration: The Trk to cancer. Cancer Treat. Res. 2003, 115, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Busam, K.J.; Benayed, R.; Cimera, R.; Wang, J.; Denley, R.; Rao, M.; Aryeequaye, R.; Mullaney, K.; Cao, L.; et al. Identification of NTRK3 Fusions in Childhood Melanocytic Neoplasms. J. Mol. Diagn. 2017, 19, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Barnhill, R.L.; Lee, S.; Li, Y.; Shao, Y.; Easton, J.; Dalton, J.; Zhang, J.; Pappo, A.; Bahrami, A. The landscape of fusion transcripts in spitzoid melanoma and biologically indeterminate spitzoid tumors by RNA sequencing. Mod. Pathol. 2016, 29, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VandenBoom, T.; Quan, V.L.; Zhang, B.; Garfield, E.M.; Kong, B.Y.; Isales, M.C.; Panah, E.; Igartua, C.; Taxter, T.; Beaubier, N.; et al. Genomic Fusions in Pigmented Spindle Cell Nevus of Reed. Am. J. Surg. Pathol. 2018, 42, 1042–1051. [Google Scholar] [CrossRef]
- Lee, C.Y.; Sholl, L.M.; Zhang, B.; Merkel, E.A.; Amin, S.M.; Guitart, J.; Gerami, P. Atypical Spitzoid Neoplasms in Childhood: A Molecular and Outcome Study. Am. J. Dermatopathol. 2017, 39, 181–186. [Google Scholar] [CrossRef]
- Zarabi, S.K.; Azzato, E.M.; Tu, Z.J.; Ni, Y.; Billings, S.D.; Arbesman, J.; Funchain, P.; Gastman, B.; Farkas, D.H.; Ko, J.S. Targeted next generation sequencing (NGS) to classify melanocytic neoplasms. J. Cutan. Pathol. 2020, 47, 691–704. [Google Scholar] [CrossRef]
- Cappellesso, R.; Nozzoli, F.; Zito Marino, F.; Simi, S.; Castiglione, F.; De Giorgi, V.; Cota, C.; Senetta, R.; Scognamiglio, G.; Anniciello, A.M.; et al. NTRK Gene Fusion Detection in Atypical Spitz Tumors. Int. J. Mol. Sci. 2021, 22, 2332. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.Y.; et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Bourhis, A.; Redoulez, G.; Quintin-Roué, I.; Marcorelles, P.; Uguen, A. Screening for NTRK-rearranged Tumors Using Immunohistochemistry: Comparison of 2 Different pan-TRK Clones in Melanoma Samples. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 194–196. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Benayed, R.; Hyman, D.M.; Drilon, A.; Zehir, A.; Frosina, D.; Arcila, M.E.; Dogan, S.; Klimstra, D.S.; Ladanyi, M.; et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am. J. Surg. Pathol. 2017, 41, 1547–1551. [Google Scholar] [CrossRef]
- Šekoranja, D.; Pižem, J.; Luzar, B. An Update on Molecular Genetic Aberrations in Spitz Melanocytic Proliferations: Correlation with Morphological Features and Biological Behavior. Acta Med. Acad. 2021, 50, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Compres, E.V.; Zhang, B.; Khan, A.U.; Sunshine, J.C.; Quan, V.L.; Gerami, P. A Series of RET Fusion Spitz Neoplasms with Plaque-Like Silhouette and Dyscohesive Nesting of Epithelioid Melanocytes. Am. J. Dermatopathol. 2021, 43, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chang, M.T.; McKay, D.; Na, N.; Zhou, B.; Yaeger, R.; Torres, N.M.; Muniz, K.; Drosten, M.; Barbacid, M.; et al. Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov. 2018, 8, 648–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, D.; Troppmair, J.; Rapp, U.R. Cot protooncoprotein activates the dual specificity kinases MEK-1 and SEK-1 and induces differentiation of PC12 cells. Oncogene 1999, 18, 1391–1400. [Google Scholar] [CrossRef] [Green Version]
- Salmeron, A.; Ahmad, T.B.; Carlile, G.W.; Pappin, D.; Narsimhan, R.P.; Ley, S.C. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 1996, 15, 817–826. [Google Scholar] [CrossRef]
- Gándara, M.L.; López, P.; Hernando, R.; Castaño, J.G.; Alemany, S. The COOH-terminal domain of wild-type Cot regulates its stability and kinase specific activity. Mol. Cell Biol. 2003, 23, 7377–7390. [Google Scholar] [CrossRef] [Green Version]
- Ceci, J.D.; Patriotis, C.P.; Tsatsanis, C.; Makris, A.M.; Kovatch, R.; Swing, D.A.; Jenkins, N.A.; Tsichlis, P.N.; Copeland, N.G. Tpl-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev. 1997, 11, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Newman, S.; Pappo, A.; Raimondi, S.; Zhang, J.; Barnhill, R.; Bahrami, A. Pathologic Characteristics of Spitz Melanoma With MAP3K8 Fusion or Truncation in a Pediatric Cohort. Am. J. Surg. Pathol. 2019, 43, 1631–1637. [Google Scholar] [CrossRef]
- Aramini, J.M.; Vorobiev, S.M.; Tuberty, L.M.; Janjua, H.; Campbell, E.T.; Seetharaman, J.; Su, M.; Huang, Y.J.; Acton, T.B.; Xiao, R.; et al. The RAS-Binding Domain of Human BRAF Protein Serine/Threonine Kinase Exhibits Allosteric Conformational Changes upon Binding HRAS. Structure 2015, 23, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Richtig, G.; Hoeller, C.; Kashofer, K.; Aigelsreiter, A.; Heinemann, A.; Kwong, L.N.; Pichler, M.; Richtig, E. Beyond the BRAF. Br. J. Dermatol. 2017, 177, 936–944. [Google Scholar] [CrossRef]
- Hutchinson, K.E.; Lipson, D.; Stephens, P.J.; Otto, G.; Lehmann, B.D.; Lyle, P.L.; Vnencak-Jones, C.L.; Ross, J.S.; Pietenpol, J.A.; Sosman, J.A.; et al. BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin. Cancer Res. 2013, 19, 6696–6702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.W.; Kim, S.M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Khan, A.U.; Compres, E.V.; Zhang, B.; Sunshine, J.C.; Quan, V.L.; Gerami, P. BRAF fusion Spitz neoplasms; clinical morphological, and genomic findings in six cases. J. Cutan. Pathol. 2020, 47, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Perron, E.; Pissaloux, D.; Neub, A.; Hohl, D.; Tartar, M.D.; Mortier, L.; Alberti, L.; de la Fouchardiere, A. Unclassified sclerosing malignant melanomas with AKAP9-BRAF gene fusion: A report of two cases and review of BRAF fusions in melanocytic tumors. Virchows Arch. 2018, 472, 469–476. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spitz Nevus | Atypical Spitz Tumor | Malignant Spitz Tumor | |
|---|---|---|---|
| Clinical Features | |||
| Age | Mean and median age: 21 years (range 2–69 years) | Can occur at any age; more common in younger patients (<40 years) | Can occur at any age (often >40 years) |
| Location | Most commonly affects the extremities | Occurs on extremities, trunk | Occurs on extremities, trunk |
| Description | Pink or reddish plaque, papule, or nodule. | Plaque or nodule Color variegation | Enlarged Plaque or nodule Color variegation Asymmetry Evolving lesion |
| Histological Features | |||
| Size | ≤5 mm | 5–10 mm | >5 mm (often >10 mm) |
| Silhouette | Symmetric | Symmetric or asymmetric | Often asymmetric |
| Circumscription | Sharp | Often poor | Poor |
| Ulceration | Absent | Possible | Often present |
| Epidermis | Hyperplastic | Often effaced | Often effaced |
| Nesting | Vertically oriented with clefting | Irregular | Irregular and confluent |
| Pagetoid spread | Sometimes central and focal | Sometimes diffuse | Extensive |
| Maturation | Present | Sometimes partial or absent | Absent |
| Necrosis | Absent | Usually absent | Sometimes present |
| Kamino bodies | Present | Often absent | Absent |
| Deep margin | Pushing | Mostly pushing | Often infiltrative |
| Inflammation | Inconspicuous | Patchy | Patchy or band-like |
| Cytological Features | |||
| Shape | Enlarged epithelioid or spindle cells | Enlarged epithelioid or spindle cells with increasing atypia | Enlarged epithelioid or spindle cells with marked atypia |
| Pleomorphism | Absent or mild | Mild to severe | Moderate to severe |
| Cytoplasm | Ground glass | Granular | Granular |
| Nucleus | Finely dispersed chromatin | Heterogeneous chromatin | Hyperchromasia |
| Nucleolus | Distinct | Increasingly prominent | Large |
| Nuclear/cytoplasmic ratio | Low | Intermediate to high | High |
| Pigment | Superficial distribution | Variable distribution | Variable, often irregular distribution |
| Mitotic rate | 0–2/mm2 | 2–6 mitoses/mm2 | 2–6 mitoses/mm2 (often > 6 mitoses/mm2) |
| Atypical mitoses | Absent | Mostly absent | Present |
| Immunohistochemical Features | |||
| HMB45 | Diminished with depth in dermal component | Diminished or variable with depth in dermal component | Deep staining common |
| Ki-67 | <5% | 5–15% | >15% |
| p16 | Present (checkerboard pattern) | Sometimes diminished or absent | Often diminished or absent |
| Molecular Features | |||
| CGH array | Isolated gains of 7p and 11q, tetraploidy | Often > 1 chromosomal abnormality Gains of 6p25 | Often > 1 chromosomal abnormality Gains of 6p25 |
| Loss of 9p21 | Absent | Sometimes present (heterozygous or homozygous) | Often present (homosygous) |
| TP53 mutations | Absent | Sometimes present | Often present |
| TERT promoter mutations | Absent | Sometimes present | Often present |
| Histological Features | Driver Alteration | Immunohistochemistry | Molecular Analyses |
|---|---|---|---|
| Symmetric plaque-like lesion Infiltrative borders Epithelioid and spindled large melanocytes Low grade cytological atypia Low mitotic rate Desmoplastic stromal reaction Predominantly intradermal growth | HRAS mutations 11p gains | HRASQ61R (clone SP174) not useful HRASWT | NGS CGH or FISH |
| Symmetrical dome/wedge-shaped large lesion Epithelioid and spindle melanocytes Mild to moderate cytological atypia Low mitotic rate Plexiform growth pattern Absent or scant pigmentation Absent or scant Kamino bodies | ALK fusions | ALK (clones D5F3 and 5A4) | FISH or NGS |
| Plaque-like or nodular lesion Epithelioid and spindled melanocytes Mild to moderate cytological atypia Low mitotic rate Prominent junctional component Transepidermal elimination of nests Adnexal involvement Numerous Kamino bodies | ROS1 fusions | ROS1 (clone D4D6) | FISH or NGS |
| Lobulated nests Rosette-like structures Epithelioid and spindled melanocytes Mild to moderate cytological atypia Low mitotic rate Extreme maturation Filigree-like rete ridges Predominantly junctional proliferation Numerous Kamino bodies | NTRK1 fusions | Pan-TRK (clone EPR17341) | NGS (FISH suggested if pan-TRK is positive but NGS is negative) |
| Pattern ETV6-related: Large coalescing and lobulated nests Epithelioid melanocytes Pleomorphic nuclei Pattern MYO5A-related: Spindle melanocytes Fascicular to plexiform growth pattern Palisading and rosettes-like structures Pattern MYO5A-related: Epithelioid melanocytes Syncytial arrangement Central desmoplastic stroma Peripheral collagen trapping | NTRK3 fusions | Pan-TRK (clone EPR17341) | NGS (FISH suggested if pan-TRK is positive but NGS is negative) |
| Symmetrical, well-circumscribed proliferation with plaque-like silhouette Small to intermediate-sized epithelioid and Spindle melanocytes Low grade cytological atypia Nested growth | RET fusions | Not available | FISH or NGS |
| Symmetric dome-shape lesion Small to intermediate-sized epithelioid and spindle melanocytes Low grade cytological atypia Nested growth | MET fusions | Not available | FISH or NGS |
| Penetrating nevus/dysplastic nevus-like architecture Infiltrative margins Large epithelioid cells with relatively high degree of cito-nuclear atypia Poor maturation Lack of epidermal hyperplasia Stromal accumulation of melanophages Plexiform growing pattern Hyperpigmentation Absent or scant Kamino bodies | MAP2K1 mutations | Not available | NGS |
| Dome-shaped or nodular lesion Predominantly nested junctional component Ulceration Lack of maturation Epithelioid melanocytes Moderate to high grade cytological atypia High mitotic rate Giant multinucleated melanocytes | MAP3K8 fusions | Not available | FISH or NGS |
| Superficial dermal sheet-like architecture Basal desmoplastic stromal reaction Lack of maturation Epithelioid morphology Moderate to high grade cytological atypia High mitotic rate | BRAF fusions | Not available | FISH or NGS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dal Pozzo, C.A.; Cappellesso, R. The Morpho-Molecular Landscape of Spitz Neoplasms. Int. J. Mol. Sci. 2022, 23, 4211. https://doi.org/10.3390/ijms23084211
Dal Pozzo CA, Cappellesso R. The Morpho-Molecular Landscape of Spitz Neoplasms. International Journal of Molecular Sciences. 2022; 23(8):4211. https://doi.org/10.3390/ijms23084211
Chicago/Turabian StyleDal Pozzo, Carlo Alberto, and Rocco Cappellesso. 2022. "The Morpho-Molecular Landscape of Spitz Neoplasms" International Journal of Molecular Sciences 23, no. 8: 4211. https://doi.org/10.3390/ijms23084211