
Bioinformatics of Differentially Expressed Genes in Phorbol 12-Myristate 13-Acetate-Induced Megakaryocytic Differentiation of K562 Cells by Microarray Analysis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
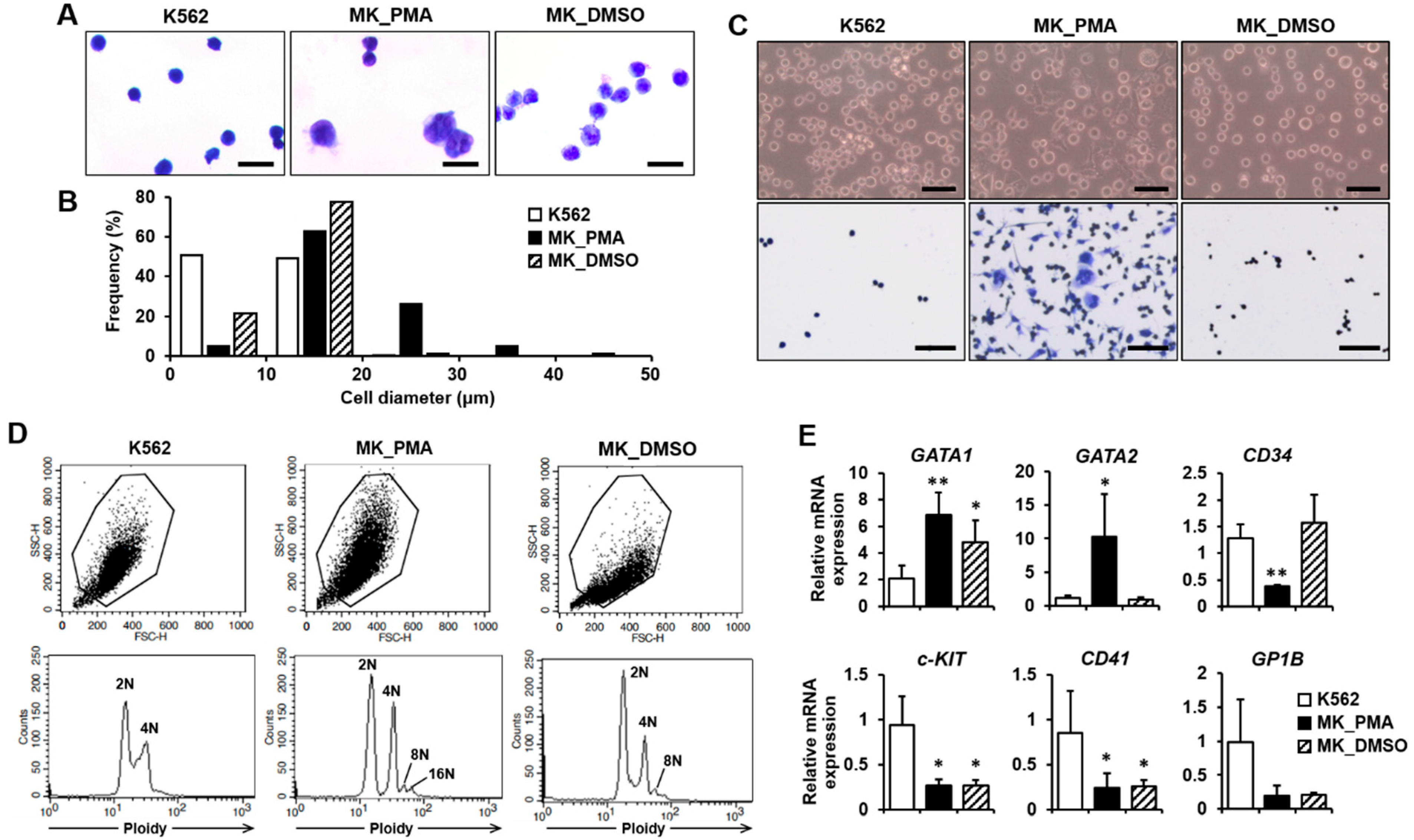
2.1. Differentiation of K562 Cells into Megakaryocytes
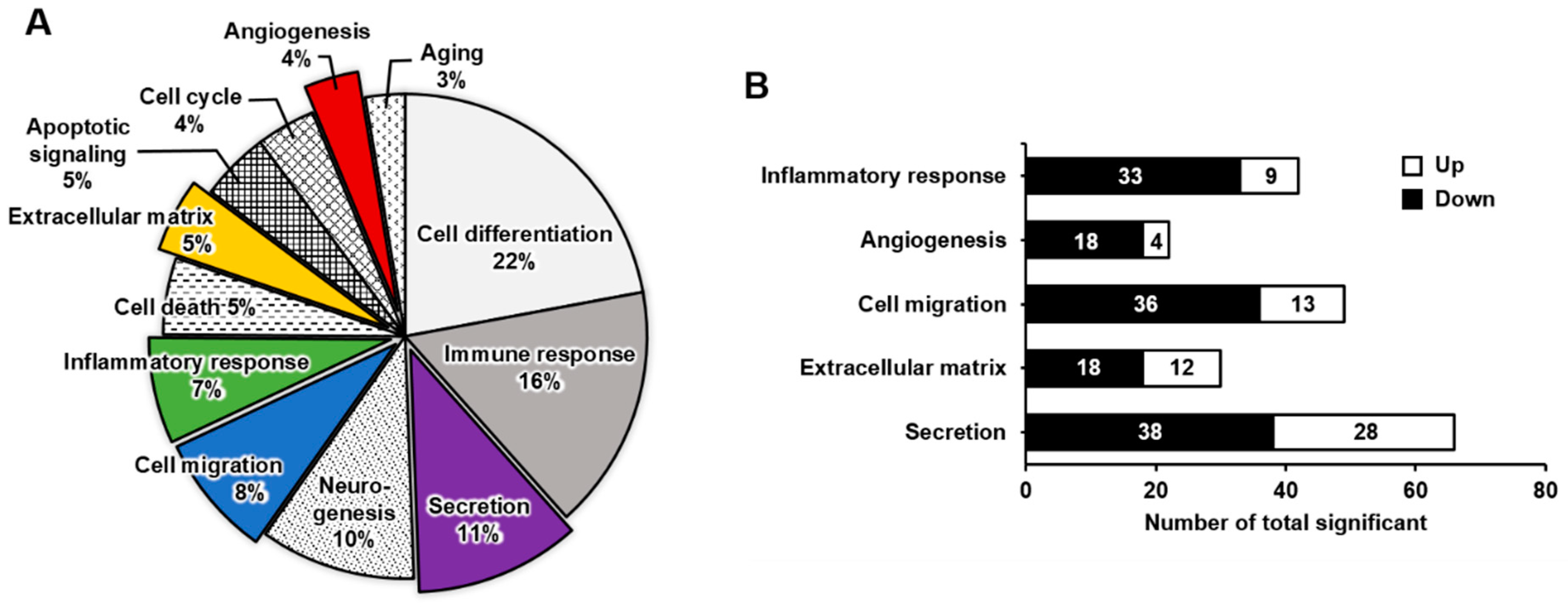
2.2. Functional Enrichment Analysis of DEGs in Mature Megakaryocytes
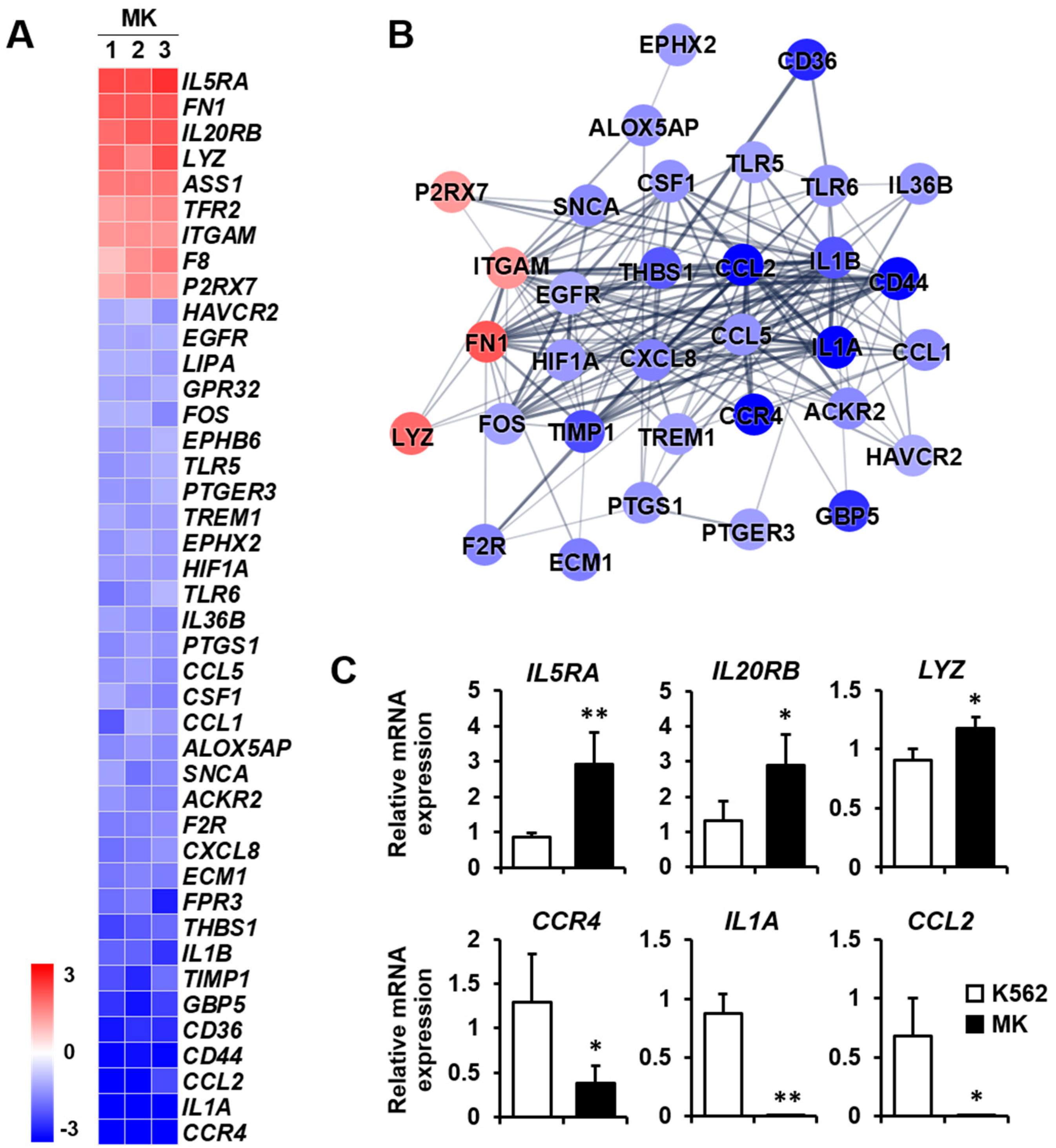
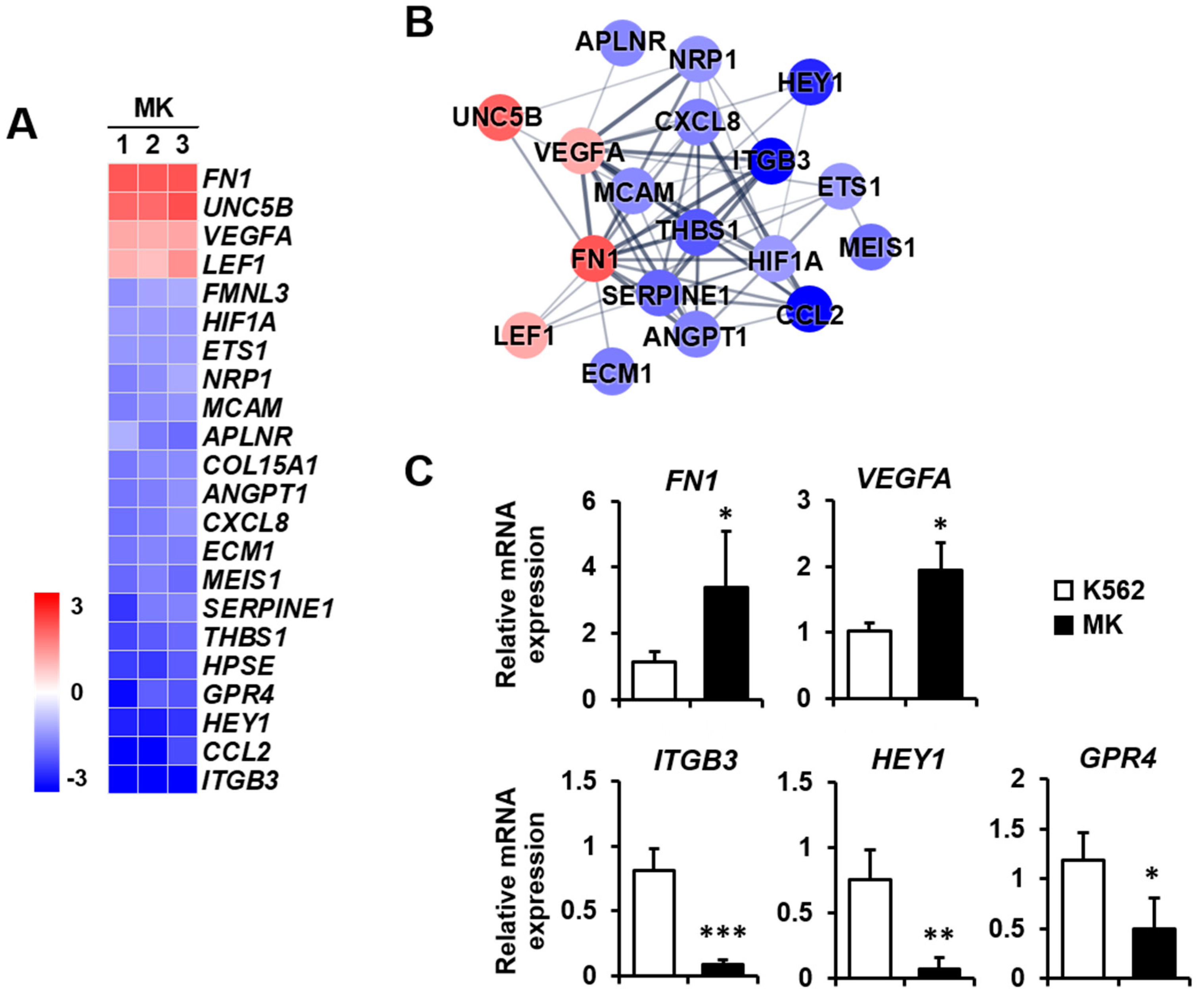
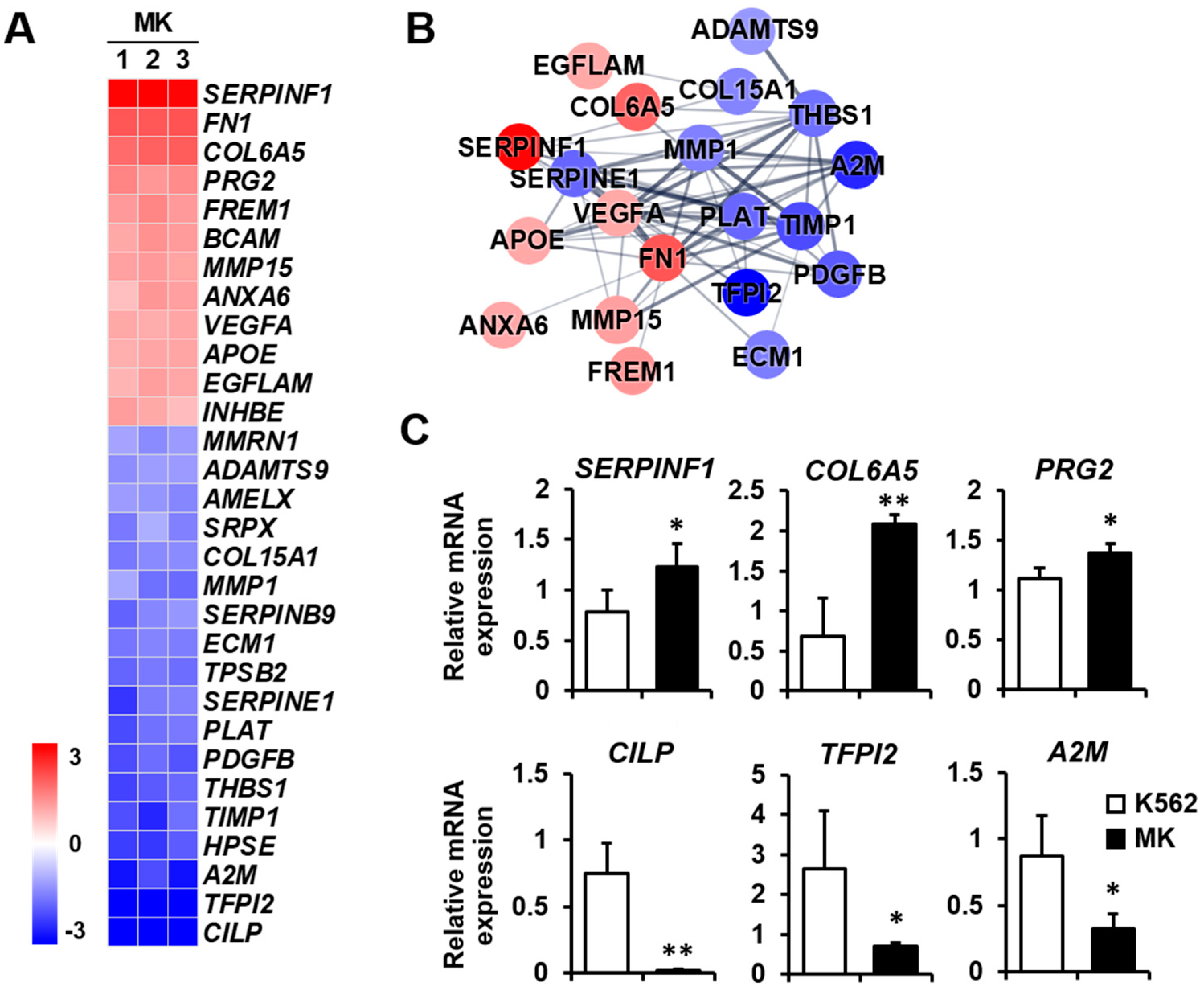
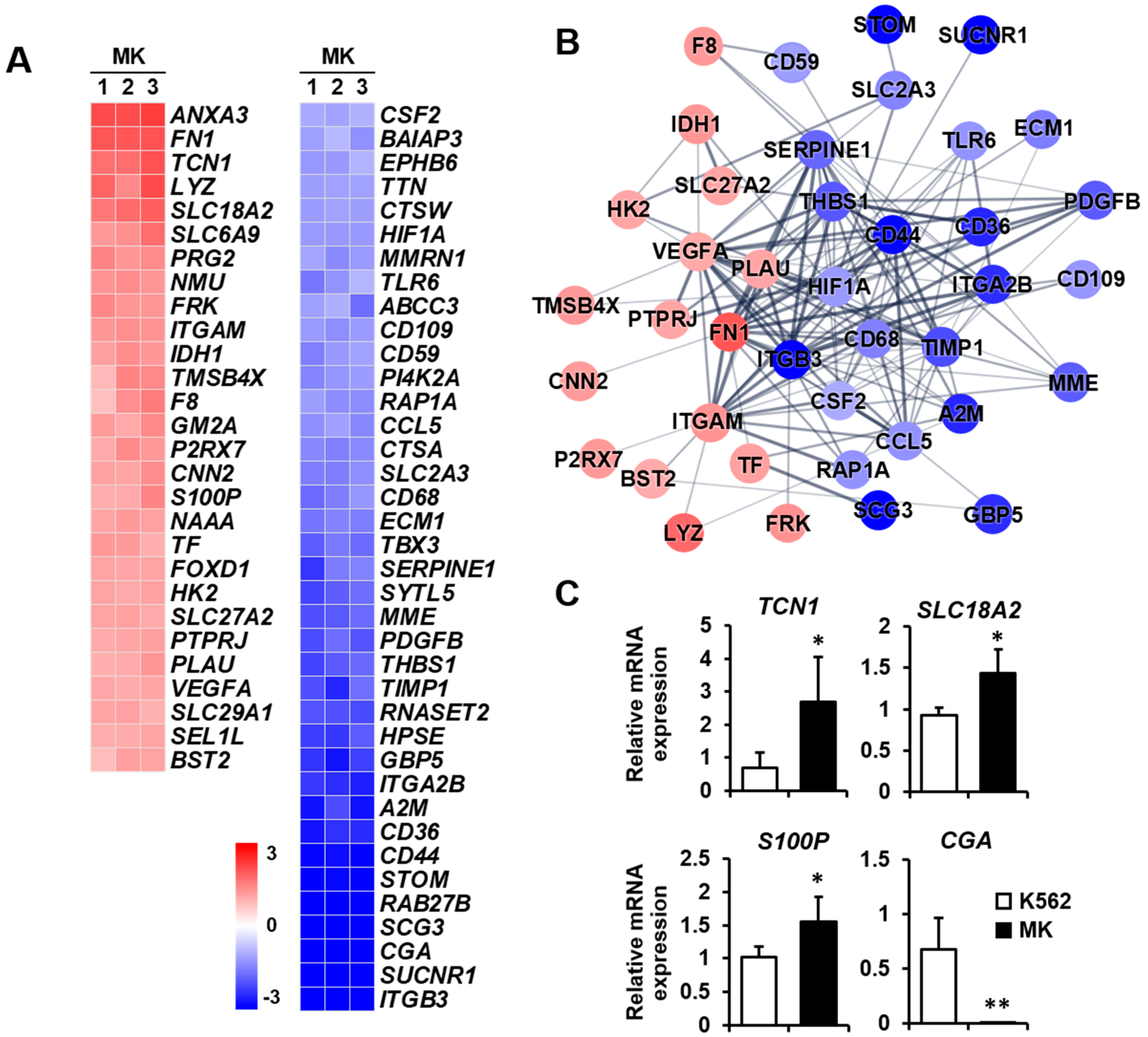
2.3. DEG Profiles and Protein–Protein Network Construction in Mature Megakaryocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Morphology and Polyploidy Analysis
4.3. RNA Extraction and qPCR
4.4. Microarray
4.5. STRING Database Analysis
4.6. Statistical Analysis
References
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A2M | alpha-2-macroglobulin | IL1B | interleukin 1 beta |
| ABCC3 | ATP binding cassette subfamily C member 3 | IL36B | interleukin 36 beta |
| ACKR2 | atypical chemokine receptor 2 | IL5RA | interleukin 5 receptor subunit alpha |
| ADAMTS9 | ADAM metallopeptidase with thrombospondin type 1 motif 9 | IL20RB | interleukin 20 receptor subunit beta |
| INHBE | inhibin subunit beta E | ||
| ALOX5AP | arachidonate 5-lipoxygenase activating protein | ITGA2 | integrin subunit alpha 2 |
| AMELX | amelogenin X-linked | ITGA9 | integrin subunit alpha 9 |
| ANGPT1 | angiopoietin 1 | ITGA2B | integrin subunit alpha 2b |
| ANXA3 | annexin A3 | ITGAM | integrin subunit alpha M |
| ANXA6 | annexin A6 | ITGB3 | integrin beta 3 |
| APLNR | apelin receptor | ITGB5 | integrin subunit beta 5 |
| APOE | apolipoprotein E | LEF1 | lymphoid enhancer-binding factor 1 |
| ARID5B | AT-rich interaction domain 5B | LIPA | lipase A, lysosomal acid type |
| ASS1 | argininosuccinate synthase1 | LRP12 | LDL receptor related protein 12 |
| BAIAP3 | BAI1 associated protein 3 | LYZ | lysozyme |
| BAMBI | BMP and activin membrane bound inhibitor | MAP1B | microtubule-associated protein 1B |
| BCAM | basal cell adhesion molecule | MCAM | melanoma cell adhesion molecule |
| BST2 | bone marrow stromal cell antigen 2 | MEIS1 | meis homeobox 1 |
| BVES | blood vessel epicardial substance | MME | membrane metalloendopeptidase |
| c-KIT | a receptor tyrosine kinase coded by the KIT gene | MMP1 | matrix metallopeptidase 1 |
| CCL1 | C-C motif chemokine ligand 1 | MMP15 | matrix metallopeptidase 15 |
| CCL2 | C-C motif chemokine ligand 2 | MMRN1 | multimerin 1 |
| CCL5 | C-C motif chemokine ligand 5 | NAAA | N-acylethanolamine acid amidase |
| CCR4 | C-C motif chemokine receptor 4 | NF-E2 | nuclear factor-erythroid 2 |
| CD36 | CD36 molecule | NMU | neuromedin U |
| CD44 | CD44 molecule | NR4A2 | nuclear receptor subfamily 4 group A member 2 |
| CD59 | CD59 molecule | NRP1 | neuropilin1 |
| CD68 | CD68 molecule | P2RX7 | >purinergic receptor P2X 7 |
| CD84 | CD84 molecule | PDGFB | platelet derived growth factor subunit B |
| CD109 | CD109 molecule | PDXDC2P | pyridoxal dependent decarboxylase domain containing 2, pseudogene |
| CGA | glycoprotein hormones alpha polypeptide | ||
| CILP | cartilage intermediate layer protein | PI4K2A | phosphatidylinositol 4-kinase type 2 alpha |
| CNN2 | calponin 2 | PLAT | plasminogen activator, tissue type |
| COL6A5 | collagen type VI alpha 5 chain | PLAU | plasminogen activator, urokinase |
| COL15A1 | collagen type XV alpha 1 chain | PRG2 | proteoglycan2, pro eosinophil major basic protein |
| CSF1 | colony stimulating factor 1 | PSTPIP2 | proline-serine-threonine phosphatase interacting protein2 |
| CSF2 | colony stimulating factor 2 | ||
| CTSA | cathepsin A | PTGER3 | prostaglandin E receptor 3 |
| CTSW | cathepsin W | PTGS1 | prostaglandin-endoperoxide synthase 1 |
| CXCL8 | C-X-C motif chemokine ligand 8 | PTPRJ | protein tyrosine phosphatase receptor type J |
| CXCL16 | C-X-C motif chemokine ligand 16 | RAB27B | RAS oncogene family member |
| DDIT4 | DNA damage inducible transcript 4 | RAP1A | RAP1A, member of RAS oncogene family |
| ECM1 | extracellular matrix protein 1 | RHOBTB1 | Rho related BTB domain containing 1 |
| EGFLAM | EGF like, fibronectin type III and laminin G domains | RNASET2 | ribonuclease T2 |
| EGFR | epidermal growth factor receptor | S100P | S100 calcium-binding protein P |
| EPHB6 | EPH receptor B6 | SCG3 | secretogranin III |
| EPHX2 | epoxide hydrolase2 | SEL1L | SEL1L adaptor subunit of ERAD E3 ubiquitin ligase |
| ESAM | endothelial cell adhesion molecule | SELPLG | selectin P ligand |
| ETS1 | ETS proto-oncogene1, transcription factor | SEMA3C | semaphorin 3C |
| FPR3 | formyl peptide receptor 3 | SERPINB9 | serpin family B member 9 |
| F2R | coagulation factor II thrombin receptor | SERPINE1 | serpin family E member 1 |
| F8 | coagulation factor VIII | SERPINF1 | serpin peptidase inhibitor clade F member 1 |
| FMNL3 | formin like 3 | SLC2A3 | solute carrier family 2 member 3 |
| FN1 | fibronectin 1 | SLC6A9 | solute carrier family 6 member 9 |
| FOS | Fos proto-oncogene, AP-1 transcription factor subunit | SLC18A2 | solute carrier family 18 member A2 |
| FOXD1 | forkhead box D1 | SLC27A2 | solute carrier family 27 member 2 |
| FREM1 | FRAS1 related extracellular matrix 1 | SLC29A1 | solute carrier family 29 member 1 |
| FRK | Fyn related Src family tyrosine kinase | SNCA | synuclein alpha |
| GATA1 | GATA binding factor 1 | SRPX | sushi repeat containing protein X-linked |
| GATA2 | GATA binding factor 2 | STOM | stomatin |
| GBP5 | Guanylate binding protein 5 | SUCNR1 | succinate receptor 1 |
| GM2A | GM2 ganglioside activator | SYTL5 | synaptotagmin like 5 |
| GP1B | platelet glycoprotein 1b | TBX3 | T-box transcription factor 3 |
| GPR4 | G protein-coupled receptor 4 | TCN1 | transcobalamin 1 |
| GPR32 | G protein-coupled receptor 32 | TF | transferrin |
| GPR183 | G protein-coupled receptor 183 | TFPI2 | tissue factor pathway inhibitor 2 |
| GYPA | glycophorin A (MNS blood group) | TFR2 | transferrin receptor 2 |
| HAVCR2 | hepatitis A virus cellular receptor 2 | THBS1 | thrombospondin 1 |
| HBEGF | heparin binding EGF like growth factor | TIMP1 | TIMP metallopeptidase inhibitor 1 |
| HEY1 | hes-related family bHLH transcription factor with YRPW motif 1 | TLR5 | toll like receptor 5 |
| TLR6 | toll like receptor 6 | ||
| HIF1A | hypoxia inducible factor 1 subunit alpha | TMSB4X | thymosin beta 4 X-linked |
| HK2 | hexokinase 2 | TPSB2 | tryptase beta 2 |
| HOXB9 | homeobox B9 | TREM1 | triggering receptor expressed on myeloid cells 1 |
| HPSE | heparanase | TTN | titin |
| IDH1 | isocitrate dehydrogenase 1 | UNC5B | unc-5 netrin receptor B |
| IL1A | interleukin 1 alpha | VEGFA | vascular endothelial growth factor A |
References
- Ebaugh, F.G.; Bird, R.M., Jr. The normal megakaryocyte concentration in aspirated human bone marrow. Blood 1951, 6, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Cunin, P.; Nigrovic, P.A. Megakaryocytes as immune cells. J. Leukoc. Biol. 2019, 105, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.J.; French, S.L.; Machlus, K.R. New insights into the differentiation of megakaryocytes from hematopoietic progenitors. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1288–1300. [Google Scholar] [CrossRef]
- Tavian, M.; Péault, B. Embryonic development of the human hematopoietic system. Int. J. Dev. Biol. 2005, 49, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Iseki, A.; Okamura, S.; Suzuki, S.; Nakauchi, H.; Ema, H. Functional characterization of hematopoietic stem cells in the spleen. Exp. Hematol. 2011, 39, 351–359.e3. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Levine, J.D.; Fu, Y.; Deng, B.; London, R.; Groopman, J.E.; Avraham, H. Cytokine production by primary bone marrow megakaryocytes. Blood 1994, 84, 4151–4156. [Google Scholar] [CrossRef]
- Majka, M.; Janowska-Wieczorek, A.; Ratajczak, J.; Ehrenman, K.; Pietrzkowski, Z.; Kowalska, M.A.; Gewirtz, A.M.; Emerson, S.G.; Ratajczak, M.Z. Numerous growth factors, cytokines, and chemokines are secreted by human CD34+ cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood 2001, 97, 3075–3085. [Google Scholar] [CrossRef]
- Boilard, E.; Flamand, L. The role of the megakaryocyte in immunity has gone viral. Blood 2019, 133, 2001–2002. [Google Scholar] [CrossRef]
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Saunders, J., II; Georas, S.N.; Veazey, J.; et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Investig. 2021, 131, e137377. [Google Scholar] [CrossRef]
- Carramolino, L.; Fuentes, J.; García-Andrés, C.; Azcoitia, V.; Riethmacher, D.; Torres, M. Platelets play an essential role in separating the blood and lymphatic vasculatures during embryonic angiogenesis. Circ. Res. 2010, 106, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhao, L.; Chen, H.; Yin, R.H.; Li, C.Y.; Zhan, Y.Q.; Zhang, J.H.; Ge, C.H.; Yu, M.; Yang, X.M. Megakaryocytic differentiation of K562 cells induced by PMA reduced the activity of respiratory chain complex IV. PLoS ONE 2014, 9, e96246. [Google Scholar] [CrossRef]
- Bütler, T.M.; Ziemiecki, A.; Friis, R.R. Megakaryocytic differentiation of K562 cells is associated with changes in the cytoskeletal organization and the pattern of chromatographically distinct forms of phosphotyrosyl-specific protein phosphatases. Cancer Res. 1990, 50, 6323–6329. [Google Scholar]
- Sutherland, J.A.; Turner, A.R.; Mannoni, P.; McGann, L.E.; Turc, J.M. Differentiation of K562 leukemia cells along erythroid, macrophage, and megakaryocyte lineages. J. Biol. Response Mod. 1986, 5, 250–262. [Google Scholar] [PubMed]
- Huo, X.F.; Yu, J.; Peng, H.; Du, Z.W.; Liu, X.L.; Ma, Y.N.; Zhang, X.; Zhang, Y.; Zhao, H.L.; Zhang, J.W. Differential expression changes in K562 cells during the hemin-induced erythroid differentiation and the phorbol myristate acetate (PMA)-induced megakaryocytic differentiation. Mol. Cell. Biochem. 2006, 292, 155–167. [Google Scholar] [CrossRef]
- Kim, K.W.; Kim, S.H.; Lee, E.Y.; Kim, N.D.; Kang, H.S.; Kim, H.D.; Chung, B.S.; Kang, C.D. Extracellular signal-regulated kinase/90-KDA ribosomal S6 kinase/nuclear factor-kappa B pathway mediates phorbol 12-myristate 13-acetate-induced megakaryocytic differentiation of K562 cells. J. Biol. Chem. 2001, 276, 13186–13191. [Google Scholar] [CrossRef]
- Rodriguez-Esteban, R.; Jiang, X. Differential gene expression in disease: A comparison between high-throughput studies and the literature. BMC Med. Genom. 2017, 10, 59. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Lemuth, K.; Rupp, S. Microarrays as research tools and diagnostic devices. In RNA and DNA Diagnostics; Springer: Cham, Switzerland, 2015; pp. 259–280. [Google Scholar]
- Jaksik, R.; Iwanaszko, M.; Rzeszowska-Wolny, J.; Kimmel, M. Microarray experiments and factors which affect their reliability. Biol. Direct. 2015, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Luyten, A.; Chen, J.; Liu, X.S.; Shivdasani, R.A. NF-E2, FLI1 and RUNX1 collaborate at areas of dynamic chromatin to activate transcription in mature mouse megakaryocytes. Sci. Rep. 2016, 6, 30255. [Google Scholar] [CrossRef]
- Adolfsson, J.; Månsson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.J.; Thoren, L.A.; et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef]
- Bluteau, D.; Lordier, L.; Di Stefano, A.; Chang, Y.; Raslova, H.; Debili, N.; Vainchenker, W. Regulation of megakaryocyte maturation and platelet formation. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 227–234. [Google Scholar] [CrossRef]
- Marcoux, G.; Laroche, A.; Espinoza Romero, J.; Boilard, E. Role of platelets and megakaryocytes in adaptive immunity. Platelets 2021, 32, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Coultas, L.; Chawengsaksophak, K.; Rossant, J. Endothelial cells and VEGF in vascular development. Nature 2005, 438, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Hu, M.; Xu, Y.; Chen, F.; Chen, S.; Chen, M.; Qi, Y.; Shen, M.; Wang, C.; Lu, Y.; et al. Megakaryocytes promote bone formation through coupling osteogenesis with angiogenesis by secreting TGF-beta1. Theranostics 2020, 10, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, V.R.; Tomer, A. Megakaryocyte development and platelet production. Br. J. Haematol. 2006, 134, 453–466. [Google Scholar] [CrossRef]
- Mazharian, A.; Watson, S.P.; Séverin, S. Critical role for ERK1/2 in bone marrow and fetal liver-derived primary megakaryocyte differentiation, motility, and proplatelet formation. Exp. Hematol. 2009, 37, 1238–1249.e5. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Malara, A.; Currao, M.; Gruppi, C.; Celesti, G.; Viarengo, G.; Buracchi, C.; Laghi, L.; Kaplan, D.L.; Balduini, A. Megakaryocytes contribute to the bone marrow-matrix environment by expressing fibronectin, type IV collagen, and laminin. Stem Cells 2014, 32, 926–937. [Google Scholar] [CrossRef]
- Lee, S.H.; Ihn, H.J.; Park, E.K.; Kim, J.E. S100 calcium-binding protein P secreted from megakaryocytes promotes osteoclast maturation. Int. J. Mol. Sci. 2021, 22, 6129. [Google Scholar] [CrossRef]
- Malara, A.; Abbonante, V.; Di Buduo, C.A.; Tozzi, L.; Currao, M.; Balduini, A. The secret life of a megakaryocyte: Emerging roles in bone marrow homeostasis control. Cell. Mol. Life Sci. 2015, 72, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Pendaries, C.; Watson, S.P.; Spalton, J.C. Methods for genetic modification of megakaryocytes and platelets. Platelets 2007, 18, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, L.B.; Schick, B.P.; Budarf, M.L.; Driscoll, D.A.; Zackai, E.H.; Cohen, A.; Konkle, B.A. Identification of a mutation in a GATA binding site of the platelet glycoprotein Ibbeta promoter resulting in the Bernard-Soulier syndrome. J. Biol. Chem. 1996, 271, 22076–22080. [Google Scholar] [CrossRef]
- Jackers, P.; Szalai, G.; Moussa, O.; Watson, D.K. Ets-dependent regulation of target gene expression during megakaryopoiesis. J. Biol. Chem. 2004, 279, 52183–52190. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, M.; Shivdasani, R.A. Expression analysis of primary mouse megakaryocyte differentiation and its application in identifying stage-specific molecular markers and a novel transcriptional target of NF-E2. Blood 2007, 109, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jin, C.; Li, Y.; Si, J.; Cui, Y.; Rondina, M.T.; Tang, F.; Wang, Q.F. Transcriptional and spatial heterogeneity of mouse megakaryocytes at single-cell resolution. Blood 2019, 134, 275. [Google Scholar] [CrossRef]
- Lentaigne, C.; Freson, K.; Laffan, M.A.; Turro, E.; Ouwehand, W.H.; BRIDGE-BPD Consortium and the ThromboGenomics Consortium. Inherited platelet disorders: Toward DNA-based diagnosis. Blood 2016, 127, 2814–2823. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Conde, I.; Pabón, D.; Jayo, A.; Lastres, P.; González-Manchón, C. Involvement of ERK1/2, p38 and PI3K in megakaryocytic differentiation of K562 cells. Eur. J. Haematol. 2010, 84, 430–440. [Google Scholar] [CrossRef]
- Supernat, A.; Popęda, M.; Pastuszak, K.; Best, M.G.; Grešner, P.; Veld, S.I.; Siek, B.; Bednarz-Knoll, N.; Rondina, M.T.; Stokowy, T.; et al. Transcriptomic landscape of blood platelets in healthy donors. Sci. Rep. 2021, 11, 15679. [Google Scholar] [CrossRef]
- Kong, C.; Zhan, B.; Piao, C.; Zhang, Z.; Zhu, Y.; Li, Q. Overexpression of UNC5B in bladder cancer cells inhibits proliferation and reduces the volume of transplantation tumors in nude mice. BMC Cancer 2016, 16, 892. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Zhang, X.; Kang, Y.; Wu, J.; Chen, J.; Li, H.; Guo, Y.; Liu, B.; Shao, Z.; Zhao, X. Genome-wide transcriptional profiling analysis reveals annexin A6 as a novel EZH2 target gene involving gastric cellular proliferation. Mol. Biosyst. 2015, 11, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.R.; Campbell, I.D. Fibronectin structure and assembly. Curr. Opin. Cell Biol. 1994, 6, 648–655. [Google Scholar] [CrossRef]
- Zhou, Y.; Shu, C.; Huang, Y. Fibronectin promotes cervical cancer tumorigenesis through activating FAK signaling pathway. J. Cell. Biochem. 2019, 120, 10988–10997. [Google Scholar] [CrossRef] [PubMed]
- Tonini, T.; Rossi, F.; Claudio, P.P. Molecular basis of angiogenesis and cancer. Oncogene 2003, 22, 6549–6556. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Feng, Q.; Tan, G. microRNA-613 exerts anti-angiogenic effect on nasopharyngeal carcinoma cells through inactivating the AKT signaling pathway by down-regulating FN1. Biosci. Rep. 2019, 39, BSR20182196. [Google Scholar] [CrossRef]
- Yang, J.G.; Wang, L.L.; Ma, D.C. Effects of vascular endothelial growth factors and their receptors on megakaryocytes and platelets and related diseases. Br. J. Haematol. 2018, 180, 321–334. [Google Scholar] [CrossRef]
- Oommen, S.; Gupta, S.K.; Vlahakis, N.E. Vascular endothelial growth factor A (VEGF-A) induces endothelial and cancer cell migration through direct binding to integrin α9β1: Identification of a specific α9β1 binding site. J. Biol. Chem. 2011, 286, 1083–1092. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kwak, M.K.; Moon, S.A.; Choi, Y.J.; Baek, J.E.; Park, S.Y.; Kim, B.J.; Lee, S.H.; Koh, J.M. Regulation of bone metabolism by megakaryocytes in a paracrine manner. Sci. Rep. 2020, 10, 2277. [Google Scholar] [CrossRef]
- Sottile, J. Regulation of angiogenesis by extracellular matrix. Biochim. Biophys. Acta 2004, 1654, 13–22. [Google Scholar] [CrossRef]
- Alford, A.I.; Kozloff, K.M.; Hankenson, K.D. Extracellular matrix networks in bone remodeling. Int. J. Biochem. Cell Biol. 2015, 65, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jiang, W.G.; Grant, M.B.; Boulton, M. Pigment epithelium-derived factor inhibits angiogenesis via regulated intracellular proteolysis of vascular endothelial growth factor receptor 1. J. Biol. Chem. 2006, 281, 3604–3613. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, N.; Tombran-Tink, J.; Niyibizi, C. Pigment epithelium-derived factor enhances differentiation and mineral deposition of human mesenchymal stem cells. Stem Cells 2013, 31, 2714–2723. [Google Scholar] [CrossRef]
- Li, F.; Armstrong, G.B.; Tombran-Tink, J.; Niyibizi, C. Pigment epithelium derived factor upregulates expression of vascular endothelial growth factor by human mesenchymal stem cells: Possible role in PEDF regulated matrix mineralization. Biochem. Biophys. Res. Commun. 2016, 478, 1106–1110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suijker, J.; Baelde, H.J.; Roelofs, H.; Cleton-Jansen, A.M.; Bovée, J.V. The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or -2 blocks osteoblast differentiation in vitro and in vivo. Oncotarget 2015, 6, 14832–14842. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.Z.; Qi, H.; Weidema, A.F.; Zhang, Q.; Panupinthu, N.; Crawford, D.T.; Grasser, W.A.; Paralkar, V.M.; Li, M.; Audoly, L.P.; et al. Deletion of the P2X7 nucleotide receptor reveals its regulatory roles in bone formation and resorption. Mol. Endocrinol. 2003, 17, 1356–1367. [Google Scholar] [CrossRef]
- Zeng, D.; Yao, P.; Zhao, H. P2X7, a critical regulator and potential target for bone and joint diseases. J. Cell. Physiol. 2019, 234, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Kim, J.G.; Kim, B.S.; Lee, J.; Pi, S.H.; Lim, H.D.; Shin, H.I.; Cho, E.S.; You, H.K. BST2 mediates osteoblast differentiation via the BMP2 signaling pathway in human alveolar-derived bone marrow stromal cells. PLoS ONE 2016, 11, e0158481. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Lorenzo, P.; Bayliss, M.T.; Heinegård, D. A novel cartilage protein (CILP) present in the mid-zone of human articular cartilage increases with age. J. Biol. Chem. 1998, 273, 23463–23468. [Google Scholar] [CrossRef]
- Liu, L.; He, J.; Liu, C.; Yang, M.; Fu, J.; Yi, J.; Ai, X.; Liu, M.; Zhuang, Y.; Zhang, Y.; et al. Cartilage intermediate layer protein affects the progression of intervertebral disc degeneration by regulating the extracellular microenvironment (Review). Int. J. Mol. Med. 2021, 47, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.N.; Cook, B.; Liu, Y.; Chilukuri, K.; Stack, M.S.; Foster, D.C.; Kisiel, W.; Woodley, D.T. HT-1080 fibrosarcoma cell matrix degradation and invasion are inhibited by the matrix-associated serine protease inhibitor TFPI-2/33 kDa MSPI. Int. J. Cancer 1998, 76, 749–756. [Google Scholar] [CrossRef]
- Konduri, S.D.; Rao, C.N.; Chandrasekar, N.; Tasiou, A.; Mohanam, S.; Kin, Y.; Lakka, S.S.; Dinh, D.; Olivero, W.C.; Gujrati, M.; et al. A novel function of tissue factor pathway inhibitor-2 (TFPI-2) in human glioma invasion. Oncogene 2001, 20, 6938–6945. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.P.; Creasey, A.A.; Chang, A.; Abbink, J.J.; Roem, D.; Eerenberg, A.J.; Hack, C.E.; Taylor, F.B., Jr. Alpha-2-macroglobulin functions as an inhibitor of fibrinolytic, clotting, and neutrophilic proteinases in sepsis: Studies using a baboon model. Infect. Immun. 1993, 61, 5035–5043. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Park, N.R.; Kim, J.-E. Bioinformatics of Differentially Expressed Genes in Phorbol 12-Myristate 13-Acetate-Induced Megakaryocytic Differentiation of K562 Cells by Microarray Analysis. Int. J. Mol. Sci. 2022, 23, 4221. https://doi.org/10.3390/ijms23084221
Lee S-H, Park NR, Kim J-E. Bioinformatics of Differentially Expressed Genes in Phorbol 12-Myristate 13-Acetate-Induced Megakaryocytic Differentiation of K562 Cells by Microarray Analysis. International Journal of Molecular Sciences. 2022; 23(8):4221. https://doi.org/10.3390/ijms23084221
Chicago/Turabian StyleLee, Seung-Hoon, Na Rae Park, and Jung-Eun Kim. 2022. "Bioinformatics of Differentially Expressed Genes in Phorbol 12-Myristate 13-Acetate-Induced Megakaryocytic Differentiation of K562 Cells by Microarray Analysis" International Journal of Molecular Sciences 23, no. 8: 4221. https://doi.org/10.3390/ijms23084221
APA StyleLee, S.-H., Park, N. R., & Kim, J.-E. (2022). Bioinformatics of Differentially Expressed Genes in Phorbol 12-Myristate 13-Acetate-Induced Megakaryocytic Differentiation of K562 Cells by Microarray Analysis. International Journal of Molecular Sciences, 23(8), 4221. https://doi.org/10.3390/ijms23084221