
Next-Generation Sequencing Screening of 43 Families with Non-Syndromic Early-Onset High Myopia: A Clinical and Genetic Study

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Results
3. Discussion
3.1. Genes Related to Isolated EoHM
3.2. Genes Related to Inherited Retinal Diseases
3.3. Genes Related to Vitreoretinal Inherited Diseases
3.4. Other Genes
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CODE | Description |
|---|---|
| PVS1 | Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LOF is a known mechanism of disease. |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history. |
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product. |
| BS1 | Allele frequency is greater than expected for disorder. |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation. |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants. |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before. |
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.) |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation. |
References
- Aránguez-Cortés, C.; Cortés-Orduña, I.; del-Río-Fernández, S.; Donate-López, J.; Franco-Iglesias, G.; García-Sáenz, S.; Gil-Ruiz, R.; Isasi-Saseta, M.; Martín-Justicia, A.; Martín-Hernández, E.; et al. Manual de Refracción para Residentes de Oftalmología, 2nd ed.; Essilor: Madrid, Spain, 2007. [Google Scholar]
- Durajczyk, M.; Grudzińska, E.; Obszańska, A.; Modrzejewska, M. Myopia. Definition and classification according to the latest knowledge. Ophtha Therapy 2021, 4, 226–231. [Google Scholar] [CrossRef]
- Holden, B.A.; Fricke, T.R.; Wilson, D.A.; Jong, M.; Naidoo, K.S.; Sankaridurg, P.; Wong, T.Y.; Naduvilath, T.; Resnikoff, S. Global Prevalence of Myopia and High Myopia and Temporal Trends from 2000 through 2050. Ophthalmology 2016, 123, 1036–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Xiao, X.; Li, S.; Jia, X.; Zhang, Q. Frequent mutations of RetNet genes in eoHM: Further confirmation in 325 probands and comparison with late-onset high myopia based on exome sequencing. Exp. Eye Res. 2018, 171, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Peregrina, C.C.; Sanchez-Tena, M.A.M.A.; Martinez-Perez, C.C.; Villa-Collar, C.C. Prevalence and Risk Factors of Myopia in Spain. J. Ophthalmol. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Pärssinen, O.; Kauppinen, M. Risk factors for high myopia: A 22-year follow-up study from childhood to adulthood. Acta Ophthalmol. 2018, 97, 510–518. [Google Scholar] [CrossRef]
- Czepita, D.; Gosławski, W.; Mojsa, A.; Muszyńska-Lachota, I. Role of light emitted by incandescent or fluorescent lamps in the development of myopia and astigmatism. Med. Sci. Monit. 2004, 10, CR168–CR171. [Google Scholar]
- Németh, J.; Tapasztó, B.; Aclimandos, W.A.; Kestelyn, P.; Jonas, J.B.; De Faber, J.T.H.; Januleviciene, I.; Grzybowski, A.; Nagy, Z.Z.; Parssinen, O.; et al. Update and guidance on management of myopia. European Society of Ophthalmology in cooperation with International Myopia Institute. Eur. J. Ophthalmol. 2021, 31, 853–883. [Google Scholar] [CrossRef]
- Chassine, T.; Villain, M.; Hamel, C.P.; Daien, V. How can we prevent myopia progression? Eur. J. Ophthalmol. 2015, 25, 280–285. [Google Scholar] [CrossRef]
- Liu, F.; Wang, J.; Xing, Y.; Li, T. Mutation screening of 17 candidate genes in a cohort of 67 probands with early-onset high myopia. Ophthalmic Physiol. Opt. 2020, 40, 271–280. [Google Scholar] [CrossRef]
- Swierkowska, J.; Karolak, J.A.; Gambin, T.; Rydzanicz, M.; Frajdenberg, A.; Mrugacz, M.; Podfigurna-Musielak, M.; Stankiewicz, P.; Lupski, J.R.; Gajecka, M. Variants in FLRT3 and SLC35E2B identified using exome sequencing in seven high myopia families from Central Europe. Adv. Med. Sci. 2021, 66, 192–198. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, K.; He, W.; Yang, J.; Sun, X.; Jiang, C.; Dai, J.; Lu, Y. Proinflammatory status in the aqueous humor of high myopic cataract eyes. Exp. Eye Res. 2016, 142, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Coviltir, V.; Burcel, M.; Popa-Cherecheanu, A.; Ionescu, C.; Dascalescu, D.; Potop, V.; Burcea, M. Update on Myopia Risk Factors and Microenvironmental Changes. J. Ophthalmol. 2019, 2019, 4960852. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhuang, X.; Fan, J.; Jiang, R.; Chang, Q.; Xu, G.; Yu, Z. Proinflammatory and angiogenesis-related cytokines in vitreous samples of highly myopic patients. Cytokine 2020, 137, 155308. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Miyazaki, D.; Sasaki, S.-I.; Miyake, K.-I.; Kaneda, S.; Ikeda, Y.; Baba, T.; Yamasaki, A.; Noguchi, Y.; Inoue, Y. Associations of inflammatory cytokines with choroidal neovascularization in highly myopic eyes. Retina 2015, 35, 344–350. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef]
- Marr, J.E.; Halliwell-Ewen, J.; Fisher, B.; Soler, L.; Ainsworth, J.R. Associations of high myopia in childhood. Eye 2001, 15, 70–74. [Google Scholar] [CrossRef]
- Verhoeven, V.J.; Hysi, P.G.; Wojciechowski, R.; Fan, Q.; Guggenheim, J.A.; Höhn, R.; MacGregor, S.; Hewitt, A.W.; Nag, A.; Cheng, C.-Y.; et al. Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat. Genet. 2013, 45, 314–318. [Google Scholar] [CrossRef]
- Wang, J.; Liu, F.; Song, X.; Li, T. Association of 5p15.2 and 15q14 with high myopia in Tujia and Miao Chinese populations. BMC Ophthalmol. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Li, Y.-J.; Goh, L.; Khor, C.C.; Fan, Q.; Yu, M.; Han, S.; Sim, X.; Ong, R.T.-H.; Wong, T.-Y.; Vithana, E.N.; et al. Genome-Wide Association Studies Reveal Genetic Variants in CTNND2 for High Myopia in Singapore Chinese. Ophthalmology 2011, 118, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Huang, L.; Xu, Y.; Xiao, X.; Li, S.; Jia, X.; Gao, B.; Wang, P.; Guo, X.; Zhang, Q. Exome Sequencing on 298 Probands with Early-Onset High Myopia: Approximately One-Fourth Show Potential Pathogenic Mutations in RetNet Genes. Investig. Opthalmol. Vis. Sci. 2015, 56, 8365–8372. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, A.K.; Tung, J.Y.; Do, C.B.; Hinds, D.A.; Mountain, J.L.; Francke, U.; Eriksson, N. Genome-Wide Analysis Points to Roles for Extracellular Matrix Remodeling, the Visual Cycle, and Neuronal Development in Myopia. PLoS Genet. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Verhoeven, V.J.M.; Wojciechowski, R.; Barathi, V.A.; Hysi, P.G.; Guggenheim, J.A.; Höhn, R.; Vitart, V.; Khawaja, A.P.; Yamashiro, K.; et al. Meta-analysis of gene–environment-wide association scans accounting for education level identifies additional loci for refractive error. Nat. Commun. 2016, 7, 11008. [Google Scholar] [CrossRef] [Green Version]
- Hysi, P.G.; Choquet, H.; Khawaja, A.P.; Wojciechowski, R.; Tedja, M.S.; Yin, J.; Simcoe, M.J.; Patasova, K.; Mahroo, O.A.; Thai, K.K.; et al. Meta-analysis of 542,934 subjects of European ancestry identifies new genes and mechanisms predisposing to refractive error and myopia. Nat. Genet. 2020, 52, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, C.S.; Taylor, D. Pediatric Ophthalmology and Strabismus, 4th ed.; Elsevier Health Sciences: Nottingham, UK, 2012. [Google Scholar]
- Utz, V.M.; Pfeifer, W.; Longmuir, S.Q.; Olson, R.; Wang, K.; Drack, A. Presentation of TRPM1-Associated Congenital Stationary Night Blindness in Children. JAMA Ophthalmol. 2018, 136, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Wiszniewski, W.; Lewis, R.A.; Stockton, D.W.; Peng, J.; Mardon, G.; Chen, R.; Lupski, J.R. Potential involvement of more than one locus in trait manifestation for individuals with Leber congenital amaurosis. Qual. Life Res. 2010, 129, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheck, L.; Davies, W.I.L.; Moradi, P.; Robson, A.G.; Kumaran, N.; Liasis, A.C.; Webster, A.R.; Moore, A.T.; Michaelides, M. Leber Congenital Amaurosis Associated with Mutations in CEP290, Clinical Phenotype, and Natural History in Preparation for Trials of Novel Therapies. Ophthalmology 2018, 125, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallum, J.M.F.; Motta, F.L.; Arno, G.; Porto, F.B.O.; Resende, R.G.; Belfort Jr, R. Clinical and molecular findings in a cohort of 152 Brazilian severe early onset inherited retinal dystrophy patients. Am. J. Med. Genet. 2020, 184, 728–752. [Google Scholar] [CrossRef]
- Kondo, H.; Matsushita, I.; Nagata, T.; Hayashi, T.; Kakinoki, M.; Uchio, E.; Kondo, M.; Ohji, M.; Kusaka, S. Novel mutations in the COL2A1 gene in Japanese patients with Stickler syndrome. Hum. Genome Var. 2016, 3, 16018. [Google Scholar] [CrossRef]
- Maddirevula, S.; Alsahli, S.; Alhabeeb, L.; Patel, N.; Alzahrani, F.; Shamseldin, H.E.; Anazi, S.; Ewida, N.; Alsaif, H.S.; Mohamed, J.Y.; et al. Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 2018, 20, 1609–1616. [Google Scholar] [CrossRef]
- Stewart, J.D.; Hudson, G.; Yu-Wai-Man, P.; Blakeley, E.L.; He, L.; Horvath, R.; Maddison, P.; Wright, A.; Griffiths, P.G.; Turnbull, D.M.; et al. OPA1 in multiple mitochondrial DNA deletion disorders. Neurology 2008, 71, 1829–1831. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Trenell, M.I.; Hollingsworth, K.G.; Griffiths, P.G.; Chinnery, P.F. OPA1 mutations impair mitochondrial function in both pure and complicated dominant optic atrophy. Brain 2010, 134, e164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouelhoda, M.; Sobahy, T.; El-Kalioby, M.; Patel, N.; Shamseldin, H.; Monies, D.; Al-Tassan, N.; Ramzan, K.; Imtiaz, F.; Shaheen, R.; et al. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet. Med. 2016, 18, 1244–1249. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Li, J.; Xiao, X.; Li, S.; Jia, X.; Sun, W.; Guo, X.; Zhang, Q. Detection of Mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 Families with Early-Onset High Myopia by Exome Sequencing. Investig. Opthalmol. Vis. Sci. 2014, 56, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran-Viet, K.-N.; Powell, C.; Barathi, V.A.; Klemm, T.; Maurer-Stroh, S.; Limviphuvadh, V.; Soler, V.; Ho, C.; Yanovitch, T.; Schneider, G.; et al. Mutations in SCO2 Are Associated with Autosomal-Dominant High-Grade Myopia. Am. J. Hum. Genet. 2013, 92, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheu-Grau, D.; Bareth, B.; Dudek, J.; Juris, L.; Vögtle, F.-N.; Wissel, M.; Leary, S.C.; Dennerlein, S.; Rehling, P.; Deckers, M. Cooperation between COA6 and SCO2 in COX2 Maturation during Cytochrome c Oxidase Assembly Links Two Mitochondrial Cardiomyopathies. Cell Metab. 2015, 21, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kars, M.E.; Başak, A.N.; Onat, O.E.; Bilguvar, K.; Choi, J.; Itan, Y.; Çağlar, C.; Palvadeau, R.; Casanova, J.-L.; Cooper, D.N.; et al. The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wakazono, T.; Miyake, M.; Yamashiro, K.; Yoshikawa, M.; Yoshimura, N. Association between SCO2 mutation and extreme myopia in Japanese patients. Jpn. J. Ophthalmol. 2016, 60, 319–325. [Google Scholar] [CrossRef]
- Zheng, Y.-H.; Cai, X.-B.; Xia, L.-Q.; Zhou, F.Y.; Wen, X.-R.; Chen, D.-F.; Han, F.; Zhou, K.J.; Jin, Z.-B.; Zhuang, W.-J.; et al. Mutational screening of AGRN, SLC39A5, SCO2, P4HA2, BSG, ZNF644, and CPSF1 in a Chinese cohort of 103 patients with nonsyndromic high myopia. Mol. Vis. 2021, 27, 706. [Google Scholar]
- Piekutowska-Abramczuk, D.; Kocyła-Karczmarewicz, B.; Małkowska, M.; Łuczak, S.; Iwanicka-Pronicka, K.; Siegmund, S.; Yang, H.; Wen, Q.; Hoang, Q.V.; Silverman, R.H.; et al. No Evidence for Association of SCO2 Heterozygosity with High-Grade Myopia or Other Diseases with Possible Mitochondrial Dysfunction. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Shi, Y.; Li, Y.; Zhang, D.; Zhang, H.; Li, Y.; Lu, F.; Liu, X.; He, F.; Gong, B.; Cai, L.; et al. Exome Sequencing Identifies ZNF644 Mutations in High Myopia. PLoS Genet. 2011, 7, e1002084. [Google Scholar] [CrossRef]
- Wang, H.; Yang, M.; Su, S.; Kang, L.; Zhu, R.; Shi, J.; Guan, H. Association of ZNF644, GRM6 and CTNND2 genes polymorphisms with high myopia. Zhonghua Yi Xue Za Zhi 2014, 94, 1289–1293. [Google Scholar] [PubMed]
- Wutz, K.; Sauer, C.; Zrenner, E.; Lorenz, B.; Alitalo, T.; Broghammer, M.; Hergersberg, M.; De La Chapelle, A.; Weber, B.H.; Wissinger, B.; et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur. J. Hum. Genet. 2002, 10, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, U.; Méjécase, C.; Ali, S.; Moosajee, M.; Kozak, I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes 2021, 12, 171. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, Z.; Xie, S.; Li, C.; Lv, L.; Zhang, M.; Zhao, J. Genetic and phenotypic characteristics of four Chinese families with fundus albipunctatus. Sci. Rep. 2017, 7, 46285. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Abdalla, E.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.; D’Angelo, R.; Sidoti, A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. Int. J. Mol. Sci. 2021, 22, 3484. [Google Scholar] [CrossRef]
- Kloss, B.A.; Tompson, S.W.; Whisenhunt, K.; Huang, S.J.; Rosenberg, T.; Young, T.L. Whole Exome Sequencing Identifies a Rare Variant Co-segregating with High Myopia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3606. [Google Scholar]
- Chen, L.; Wei, Y.; Chi, W.; Fang, D.; Jiang, X.; Zhang, S. Potential Mutations in Chinese Pathologic Myopic Patients and Contributions to Phenotype. Curr. Mol. Med. 2019, 18, 689–697. [Google Scholar] [CrossRef]
- Logan, N.S.; Gilmartin, B.; Marr, J.E.; Stevenson, M.R.; Ainsworth, J.R. Community-Based Study of the Association of High Myopia in Children with Ocular and Systemic Disease. Optom. Vis. Sci. 2004, 81, 11–13. [Google Scholar] [CrossRef] [Green Version]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, F. The Collagen Superfamily and Collagenopathies, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Sun, W.; Xiao, X.; Li, S.; Jia, X.; Zhang, Q. A novel deep intronic COL2A1 mutation in a family with early-onset high myopia/ocular-only Stickler syndrome. Ophthalmic Physiol. Opt. 2020, 40, 281–288. [Google Scholar] [CrossRef]
- Robin, N.H.; Moran, R.T.; Ala-Kokko, L. Stickler Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., et al., Eds.; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Richards, A.J.; Martin, S.; Yates, J.R.W.; Scott, J.D.; Baguley, D.M.; Pope, F.M.; Snead, M.P. COL2A1 exon 2 mutations: Relevance to the Stickler and Wagner syndromes. Br. J. Ophthalmol. 2000, 84, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Rada, J.A.; Shelton, S.; Norton, T.T. The sclera and myopia. Exp. Eye Res. 2006, 82, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Salchow, D.J.; Gehle, P. Ocular manifestations of Marfan syndrome in children and adolescents. Eur. J. Ophthalmol. 2018, 29, 38–43. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2020, 82, 100898. [Google Scholar] [CrossRef] [PubMed]
- Young, T.L.; Nallasamy, S.; Paluru, P.; Devoto, M.; Wasserman, N.; Zhou, J. Novel Locus for High–Grade Myopia in a Hutterite Population on Chromosome 10q. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2297. [Google Scholar]
- Wan, L.; Deng, B.; Wu, Z.; Chen, X. Exome sequencing study of 20 patients with high myopia. PeerJ 2018, 6, e5552. [Google Scholar] [CrossRef]
- Poll-The, B.T.; Gootjes, J.; Duran, M.; de Klerk, J.B.; Wenniger-Prick, L.J.M.D.B.; Admiraal, R.J.; Waterham, H.R.; Wanders, R.J.; Barth, P.G. Peroxisome biogenesis disorders with prolonged survival: Phenotypic expression in a cohort of 31 patients. Am. J. Med. Genet. 2004, 126, 333–338. [Google Scholar] [CrossRef]
- García-Gen, E.; Penadés, M.; Mérida, S.; Desco, C.; Araujo-Miranda, R.; Navea, A.; Bosch-Morell, F. High Myopia and the Complement System: Factor H in Myopic Maculopathy. J. Clin. Med. 2021, 10, 2600. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Wang, Y.; Li, W.; Huang, S.; Chu, X.; Wang, L.; Zhang, M.; Liu, Z. A Novel OPA1 Mutation Responsible for Autosomal Dominant Optic Atrophy with High Frequency Hearing Loss in a Chinese Family. Am. J. Ophthalmol. 2007, 143, 186–188.e1. [Google Scholar] [CrossRef]
- Li, C.; Kosmorsky, G.; Zhang, K.; Katz, B.J.; Ge, J.; Traboulsi, E.I. Optic atrophy and sensorineural hearing loss in a family caused by an R445H OPA1 mutation. Am. J. Med. Genet. 2005, 138, 208–211. [Google Scholar] [CrossRef]
- Han, J.; Thompson-Lowrey, A.J.; Reiss, A.; Mayorov, V.; Jia, H.; Biousse, V.; Newman, N.J.; Brown, M.D. OPA1 mutations and mitochondrial DNA haplotypes in autosomal dominant optic atrophy. Genet. Med. 2006, 8, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati-Bonneau, P.; Milea, D.; Bonneau, D.; Chevrollier, A.; Ferré, M.; Guillet, V.; Gueguen, N.; Loiseau, D.; de Crescenzo, M.-A.P.; Verny, C.; et al. OPA1-associated disorders: Phenotypes and pathophysiology. Int. J. Biochem. Cell Biol. 2009, 41, 1855–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBrien, N.A.; Gentle, A. Role of the sclera in the development and pathological complications of myopia. Prog. Retin. Eye Res. 2003, 22, 307–338. [Google Scholar] [CrossRef]
- Leung, K.H.; Yiu, W.C.; Yap, M.K.; Ng, P.W.; Fung, W.Y.; Sham, P.C.; Yip, S.P. Systematic investigation of the relationship between high myopia and polymorphisms of the MMP2, TIMP2, and TIMP3 genes by a DNA pooling approach. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3893–3900. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Ohno-Matsui, K.; Kojima, A.; Shimada, N.; Yasuzumi, K.; Yoshida, T.; Futagami, S.; Tokoro, T.; Mochizuki, M. Fundus Characteristics of High Myopia in Children. Jpn. J. Ophthalmol. 2005, 49, 306–311. [Google Scholar] [CrossRef]
- Gözüm, N.; Çakir, M.; Gücukoglu, A.; Sezen, F. Relationship between Retinal Lesions and Axial Length, Age and Sex in High Myopia. Eur. J. Ophthalmol. 1997, 7, 277–282. [Google Scholar] [CrossRef]
- Jagadeesh, D.; Philip, K.; Fedtke, C.; Jong, M.; Ly, A.; Sankaridurg, P. Posterior segment conditions associated with myopia and high myopia. Clin. Exp. Optom. 2020, 103, 756–765. [Google Scholar] [CrossRef]
- Nakanishi, H.; Hayashi, H.; Yamada, R.; Yamashiro, K.; Nakata, I.; Shimada, N.; Ohno-Matsui, K.; Mochizuki, M.; Ozaki, M.; Yoshitake, S.; et al. Single-Nucleotide Polymorphisms in the Promoter Region of Matrix Metalloproteinase-1, -2, and -3 in Japanese with High Myopia. Investig. Opthalmol. Vis. Sci. 2010, 51, 4432–4436. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Hsiao, Y.-W.; Zhou, J.-B. Association between MMP/TIMP Levels in the Aqueous Humor and Plasma with Axial Lengths in Myopia Patients. BioMed Res. Int. 2020, 2020, 2961742. [Google Scholar] [CrossRef] [PubMed]
- David, T.; Smye, S.; James, T.; Dabbs, T. Time-dependent stress and displacement of the eye wall tissue of the human eye. Med. Eng. Phys. 1997, 19, 131–139. [Google Scholar] [CrossRef]
- Shelton, L.; Rada, J.S. Effects of cyclic mechanical stretch on extracellular matrix synthesis by human scleral fibroblasts. Exp. Eye Res. 2007, 84, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, M.; Li, B.; Li, T.; Hu, Q.; Zhou, X. Modulation of the ERK1/2-MMP-2 pathway in the sclera of guinea pigs following induction of myopia by flickering light. Exp. Ther. Med. 2021, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schäche, M.; Baird, P.N. Assessment of the Association of Matrix Metalloproteinases with Myopia, Refractive Error and Ocular Biometric Measures in an Australian Cohort. PLoS ONE 2012, 7, e47181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, H.; Zhang, R.; Shu, Q.; Jiang, R.; Chang, Q.; Huang, X.; Jiang, C.; Xu, G. Changes of TGF-β2, MMP-2, and TIMP-2 levels in the vitreous of patients with high myopia. Graefe’s Arch. Clin. Exp. Ophthalmol. 2014, 252, 1763–1767. [Google Scholar] [CrossRef]
- Jia, Y.; Hu, D.-N.; Sun, J.; Zhou, J. Correlations Between MMPs and TIMPs Levels in Aqueous Humor from High Myopia and Cataract Patients. Curr. Eye Res. 2016, 42, 600–603. [Google Scholar] [CrossRef]
- Pan, C.W.; Cheung, C.Y.; Aung, T.; Cheung, C.M.; Zheng, Y.F.; Wu, R.Y.; Mitchell, P.; Lavanya, R.; Baskaran, M.; Wang, J.J.; et al. Differential associations of myopia with major age-related eye diseases: The Singapore Indian Eye Study. Ophthalmology 2013, 120, 284–291. [Google Scholar] [CrossRef]
- Zhu, X.; Du, Y.; Li, D.; Xu, J.; Wu, Q.; He, W.; Zhang, K.; Zhu, J.; Guo, L.; Qi, M.; et al. Aberrant TGF-β1 signaling activation by MAF underlies pathological lens growth in high myopia. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Shi, Y.; Qu, J.; Zhang, D.; Zhao, P.; Zhang, Q.; Tam, P.O.S.; Sun, L.; Zuo, X.; Zhou, X.; Xiao, X.; et al. Genetic Variants at 13q12.12 Are Associated with High Myopia in the Han Chinese Population. Am. J. Hum. Genet. 2011, 88, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Verkicharla, P.K.; Kammari, P.; Das, A.V. Myopia progression varies with age and severity of myopia. PLoS ONE 2020, 15, e0241759. [Google Scholar] [CrossRef]
- Rozema, J.; Dankert, S.; Iribarren, R.; Lanca, C.; Saw, S.-M. Axial Growth and Lens Power Loss at Myopia Onset in Singaporean Children. Investig. Opthalmol. Vis. Sci. 2019, 60, 3091–3099. [Google Scholar] [CrossRef] [Green Version]
- Mutti, D.O.; Mitchell, G.L.; Sinnott, L.T.; Jones-Jordan, L.A.; Moeschberger, M.L.; Cotter, S.A.; Kleinstein, R.N.; Manny, R.E.; Twelker, J.D.; Zadnik, K. Corneal and Crystalline Lens Dimensions Before and After Myopia Onset. Optom. Vis. Sci. 2012, 89, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, R.; Zhou, X.-T.; Lu, F.; Chen, M.; Xue, A.; Chen, S.; Qu, J. Correlation Between Myopia and Major Biometric Parameters of the Eye: A Retrospective Clinical Study. Optom. Vis. Sci. 2009, 86, E503–E508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.J.; Zhou, P.; Zhang, K.K.; Yang, J.; Luo, Y.; Lu, Y. Epigenetic regulation of αA-crystallin in high myopia-induced dark nuclear cataract. PLoS ONE 2013, 8, e81900. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, S.; Gu, J.; Guo, M.; Xia, H.; Liu, Y. UPR Activation and the Down–Regulation of α-Crystallin in Human High Myopia-Related Cataract Lens Epithelium. PLoS ONE 2015, 10, e0137582. [Google Scholar] [CrossRef]
- Han, R.; Wang, X.; Wang, D.; Wang, L.; Yuan, Z.; Ying, M.; Li, N. GPR143 Gene Mutations in Five Chinese Families with X-linked Congenital Nystagmus. Sci. Rep. 2015, 5, srep12031. [Google Scholar] [CrossRef] [Green Version]
- Vishnupriya, S.; Bindu, C.H.; Annamaneni, S.; Reddy, K.P. Association of vitamin D receptor gene start codon (Fok1) polymorphism with high myopia. Oman J. Ophthalmol. 2011, 4, 57–62. [Google Scholar] [CrossRef]
- Ramamurthy, D.; Chua, S.Y.L.; Saw, S. A review of environmental risk factors for myopia during early life, childhood and adolescence. Clin. Exp. Optom. 2015, 98, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Yao, Q.; Ma, W.; Liu, H.; Ji, J.; Li, X. Associations of vitamin D deficiency and vitamin D receptor (Cdx-2, Fok I, Bsm I and Taq I) polymorphisms with the risk of primary open-angle glaucoma. BMC Ophthalmol. 2016, 16, 116. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.Y.; Schache, M.; Ikram, M.K.; Young, T.L.; Guggenheim, J.A.; Vitart, V.; MacGregor, S.; Verhoeven, V.J.; Barathi, V.A.; Liao, J.; et al. Nine loci for ocular axial length identified through genome-wide association studies, including shared loci with refractive error. Am. J. Hum. Genet. 2013, 93, 264–277. [Google Scholar] [CrossRef] [Green Version]
- Tideman, J.W.L.; Fan, Q.; Polling, J.R.; Guo, X.; Yazar, S.; Khawaja, A.; Höhn, R.; Lu, Y.; Jaddoe, V.W.; Yamashiro, K.; et al. When do myopia genes have their effect? Comparison of genetic risks between children and adults. Genet. Epidemiol. 2016, 40, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Hou, S.; Jiang, Z.; Hu, Z.; Yang, P.; Ye, J. Association of EPHA2 polymorphisms and age-related cortical cataract in a Han Chinese population. Mol. Vis. 2011, 17, 1553–1558. [Google Scholar] [PubMed]
- Tang, S.M.; Rong, S.S.; Young, A.L.; Tam, P.O.; Pang, C.P.; Chen, L.J. PAX6 gene associated with high myopia: A meta-analysis. Optom. Vis. Sci. 2014, 91, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.M.; Ma, L.; Lu, S.Y.; Wang, Y.; Kam, K.W.; Tam, P.O.S.; Young, A.L.; Pang, C.P.; Yam, J.C.; Chen, L.J. Association of the PAX6 gene with extreme myopia rather than lower grade myopias. Br. J. Ophthalmol. 2018, 102, 570–574. [Google Scholar] [CrossRef]
- Bilbao-Malavé, V.; Recalde, S.; Bezunartea, J.; Hernandez-Sanchez, M.; González-Zamora, J.; Maestre-Rellan, L.; Ruiz-Moreno, J.M.; Araiz-Iribarren, J.; Arias, L.; Ruiz-Medrano, J.; et al. Genetic and environmental factors related to the development of myopic maculopathy in Spanish patients. PLoS ONE 2020, 15, e0236071. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-L.; Hsi, E.; Chen, K.-C.; Pan, Y.-R.; Wang, Y.-S.; Juo, S.-H.H. A Functional Polymorphism at 3′UTR of thePAX6Gene May Confer Risk for Extreme Myopia in the Chinese. Investig. Opthalmol. Vis. Sci. 2011, 52, 3500–3505. [Google Scholar] [CrossRef] [Green Version]
- Miyake, M.; Yamashiro, K.; Nakanishi, H.; Nakata, I.; Akagi-Kurashige, Y.; Tsujikawa, A.; Moriyama, M.; Ohno-Matsui, K.; Mochizuki, M.; Yamada, R.; et al. Association of paired box 6 with high myopia in Japanese. Mol. Vis. 2012, 18, 2726–2735. [Google Scholar]
- Wang, P.; Liu, X.; Ye, Z.; Gong, B.; Yang, Y.; Zhang, D.; Wu, X.; Zheng, H.; Li, Y.; Yang, Z.; et al. Association of IGF1 and IGF1R gene polymorphisms with high myopia in a Han Chinese population. Ophthalmic Genet. 2016, 38, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Penha, A.M.; Schaeffel, F.; Feldkaemper, M. Insulin, insulin-like growth factor-1, insulin receptor, and insulin-like growth factor-1 receptor expression in the chick eye and their regulation with imposed myopic or hyperopic defocus. Mol. Vis. 2011, 17, 1436–1448. [Google Scholar]
- Fedor, M.; Socha, K.; Urban, B.; Soroczyńska, J.; Matyskiela, M.; Borawska, M.H.; Bakunowicz-Łazarczyk, A. Serum concentration of zinc, copper, selenium, manganese, and Cu/Zn ratio in children and adolescents with myopia. Biol. Trace Elem. Res. 2017, 176, 1–9. [Google Scholar] [CrossRef]
- Mérida, S.; Villar, V.M.; Navea, A.; Desco, C.; Sancho-Tello, M.; Peris, C.; Bosch-Morell, F. Imbalance Between Oxidative Stress and Growth Factors in Human High Myopia. Front. Physiol. 2020, 11, 463. [Google Scholar] [CrossRef]
- Francisco, B.-M.; Salvador, M.; Amparo, N. Oxidative Stress in Myopia. Oxidative Med. Cell. Longev. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-J.; Lam, T.C.; Sze, A.Y.-H.; Li, K.-K.; Chun, R.K.-M.; Shan, S.; To, C.-H. Alteration of retinal metabolism and oxidative stress may implicate myopic eye growth: Evidence from discovery and targeted proteomics in an animal model. J. Proteom. 2020, 221, 103684. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.B.; Kim, H.K.; Hyon, J.Y.; Wee, W.R.; Shin, Y.J. Oxidative Stress Levels in Aqueous Humor from High Myopic Patients. Korean J. Ophthalmol. 2016, 30, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.L.; Heier, J.S. Choroidal Neovascularization Secondary to Myopia, Infection and Inflammation. Retin. Pharmacother. 2015, 55, 167–175. [Google Scholar] [CrossRef]
- Guía de “Manejo de las Complicaciones Retinianas en la Alta Miopía”. “Guías de Práctica Clínica de la SERV”. Available online: www.serv.es (accessed on 2 February 2022).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Hamosh, A.; Scott, A.F.; Amberger, J.; Valle, D.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM). Hum. Mutat. 1999, 15, 57–61. [Google Scholar] [CrossRef]

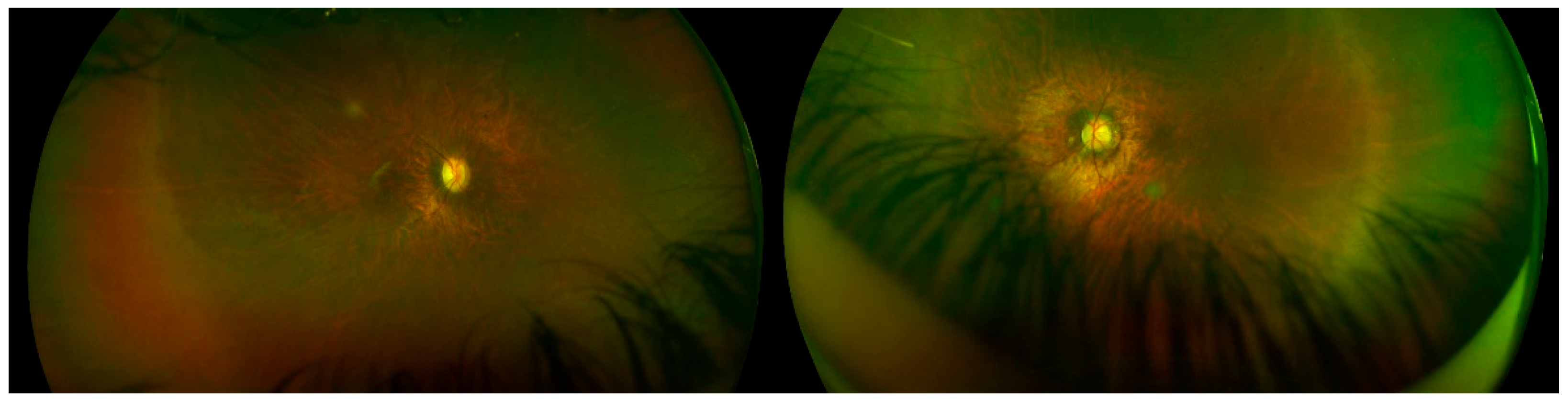





| Right Eye | Left Eye | |
|---|---|---|
| Best corrected visual acuity (decimal scale) | 0.59 ± 0.33 | 0.55 ± 0.33 |
| Axial length (mm) | 27.79 ± 2.5 | 27.95 ± 2.59 |
| Spherical refraction (diopters) | −10.8 ± 6.1 | −10.44 ± 5.38 |
| Astigmatism (diopters) | −1.71 ± 1.3 | −1.92 ± 1.4 |
| Spherical equivalent (diopters) | −11.22 ± 5.45 | −10.44 ± 4.66 |
| Family Number | Sex | BCVA OD | BCVA OS | AL OD | AL OS | Funduscopic Examination OD | Funduscopic Examination OS | SPcc OD | Astig OD | SE OD cc | SPcc OS | Astig OS | SE OS cc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFT-00074 | F | 0.6 | 0.08 | 26.6 | 26.93 | Diffuse chorioretinal atrophy, Central staphyloma | Diffuse chorioretinal atrophy, Central staphyloma | −12 | −0.5 | −12.25 | −12.75 | −2.5 | −14 |
| OFT-00155 | M | 0.125 | 0.1 | NA | NA | Healthy retina | Healthy retina | −10 | −1.25 | −10.6 | −8.75 | −2.75 | −10.1 |
| OFT-00209 | M | 0.6 | 0.7 | NA | NA | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −8.5 | −3 | −10 | −7 | −3 | −8.5 |
| OFT-00177 | F | NA | NA | NA | NA | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −24 | 0 | −24 | −18 | 0 | −18 |
| OFT-00178 | M | 0.3 | 0.4 | 26.75 | 26.65 | Healthy retina, Mild optic nerve pallor | Healthy retina | −6.75 | −4 | −8.75 | −7.25 | −3.25 | −8.88 |
| OFT-00181 | M | 0.9 | 0.9 | 26.6 | 26.7 | Healthy retina | Healthy retina | −8.25 | −1 | −8.75 | −8 | −1 | −8.5 |
| OFT-00223 | F | 0.3 | 0.3 | 28.04 | 27.62 | Atrophic optic nerve | Atrophic optic nerve | −13.5 | −2.5 | −14.75 | −13 | −0.5 | −13.25 |
| OFT-00092 * | M | 0.1 | 0.05 | NA | NA | Healthy retina | Peripheral toxoplasma scar | −0.5 | −1.5 | −1.25 | −2.25 | −0.75 | −2.6 |
| OFT-00097 | M | 0.4 | 0.2 | 26.84 | 26.47 | Tessellated fundus, Healthy optic nerve | Tessellated fundus, Healthy optic nerve | −9.75 | −5.25 | −12.35 | −10 | −5.25 | −12.6 |
| OFT-00045 | M | 0.05 | 1 | 23.56 | 23.43 | Hypopigmented fundus, Foveal hypoplasia, Colobomatous optic nerve | Hypopigmented fundus, Foveal hypoplasia | −9.75 | −2.5 | −11 | −10 | −3 | −11.5 |
| OFT-00275 | F | 0.7 | 0.1 | 27.61 | 27.6 | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −11.5 | −1 | −12 | −12 | −3.25 | −13.6 |
| OFT-00332 | M | 0.25 | 0.3 | 29.41 | 29.02 | Tessellated fundus, Epiretinal fibrosis | Tessellated fundus, WWP on inferior and temporal retina | −15.25 | −1 | −15.75 | −14.75 | −0.5 | −15 |
| OFT-00343 | F | 0.8 | 0.8 | NA | NA | Diffuse chorioretinal atrophy, Peripapillary atrophy | Diffuse chorioretinal atrophy, Peripapillary atrophy | −15.75 | −2.75 | −17.1 | −16 | −1.25 | −16.75 |
| OFT-00191 | M | 0.5 | 0.5 | 26.05 | 26.15 | Diffuse chorioretinal atrophy, Mild optic nerve pallor | Diffuse chorioretinal atrophy, Mild optic nerve pallor | −9 | −2 | −10 | −8.75 | −3.25 | −10.4 |
| OFT-00391 | M | 0.9 | NA | NA | NA | Healthy retina, WWP inferotemporal | Healthy retina | −7.25 | −2.25 | −8.375 | −7 | −3 | −8.5 |
| OFT-00407 | M | 0.6 | 0.5 | 28.26 | 27.8 | Diffuse chorioretinal atrophy, Mild optic nerve pallor | Diffuse chorioretinal atrophy, Mild optic nerve pallor | −9.75 | −3.5 | −11.5 | −9.5 | −2.5 | −10.75 |
| OFT-00429 | M | 0.8 | 0.6 | NA | NA | Diffuse chorioretinal atrophy, Peripapillary atrophy, WWP inferiorly | Diffuse chorioretinal atrophy, Peripapillary atrophy, WWP inferiorly | −20 | 0 | −20 | −19 | 0 | −19 |
| OFT-00436 | M | 0.63 | 0.3 | 27.42 | 30.93 | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −7 | −2.5 | −8.25 | −15 | −3.75 | −16.875 |
| OFT-00453 | F | NFx | Fx | NA | NA | Complete retinal detachment | Diffuse chorioretinal atrophy, Peripapillary atrophy | 2.25 | −2.25 | 1.125 | −9.5 | −1.5 | −10.25 |
| OFT-00463 | F | 0.3 | 0.05 | 32.44 | 33.57 | Severe peripapillary and macular atrophy | Severe peripapillary and macular atrophy | NA | NA | NA | NA | NA | NA |
| OFT-00474 | M | 0.1 | 0.7 | 27.43 | 25.99 | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −11.5 | −1.25 | −12.125 | −10.25 | −0.5 | −10.5 |
| OFT-00490 | F | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| OFT-00506 | F | 0.7 | 0.7 | NA | NA | Tessellated fundus | Tessellated fundus | −13.25 | −2 | −14.25 | −12.5 | −1.5 | −13.25 |
| OFT-00533 | F | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| OFT-00546 | M | 1 | 1 | 24.45 | 24.12 | Healthy retina | Healthy retina | −7.25 | −0.75 | −7.625 | −5 | −1 | −5.5 |
| OFT-00554 | M | 0.3 | 0.5 | NA | NA | Healthy retina | Healthy retina | −9 | −2.5 | −10.25 | −7.5 | −1.25 | −8.125 |
| OFT-00559 | M | 0.4 | 0.3 | NA | NA | Diffuse increase in vascular ramification, Avascular peripheral retina | Avascular peripheral retina | −7 | −1.25 | −7.625 | −7.5 | −1.75 | −8.375 |
| OFT-00568 | F | 0.16 | 0.8 | 34.09 | 33.89 | Diffuse chorioretinal atrophy, Peripapillary atrophy, Staphyloma | Diffuse chorioretinal atrophy, Peripapillary atrophy, Staphyloma | NA | NA | NA | NA | NA | NA |
| OFT-00586 | F | 0.8 | 0.05 | NA | NA | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | NA | NA | NA | NA | NA | NA |
| OFT-00590 | F | 0.63 | 0.5 | 29.39 | 28.66 | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −18.25 | −0.25 | −18.37 | −20.5 | −1.25 | −1.75 |
| OFT-00601 | M | 1 | 0.9 | 27.92 | 27.68 | Healthy retina | Healthy retina | −7 | −1 | −7.5 | −6.5 | −1 | −7 |
| OFT-00630 | F | 0.08 | 0.08 | NA | NA | Diffuse chorioretinal atrophy, Central staphyloma | Diffuse chorioretinal atrophy, Central staphyloma | −26 | NA | NA | −26 | NA | NA |
| OFT-00493 | F | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| OFT-00175 | M | 0.9 | 0.8 | 31.22 | 30.98 | Diffuse chorioretinal atrophy, Peripapillary atrophy | Diffuse chorioretinal atrophy, Peripapillary atrophy | −13.5 | −4.75 | −15.87 | −13.25 | −6 | −16.25 |
| OFT-00220 | M | 1 | 0.8 | 25.7 | 26.2 | Healthy retina | Healthy retina | −5 | −0.5 | −5.25 | −6 | −2.5 | −7.25 |
| OFT-00253 | F | 0.9 | 0.9 | 29.59 | 29.1 | Healthy retina | Healthy retina | −19.25 | 0 | −19.25 | −17.25 | −0.5 | −17.5 |
| OFT-00268 | M | 0.5 | 0.6 | 27.08 | 27.18 | Diffuse chorioretinal atrophy | Diffuse chorioretinal atrophy | −7.25 | −0.75 | −7.6 | −7 | −1 | −7.5 |
| OFT-00435 | F | 0.5 | 0.5 | 24.99 | 28.13 | Healthy retina | Healthy retina | −21.25 | −1.5 | −22 | −14 | −3.25 | −15.625 |
| OFT-00443 | M | 1.2 | 1 | NA | NA | RPE hypertrophy, WWP temporal and superior | Healthy retina | −7.75 | −0.5 | −8 | −8.5 | −0.5 | −8.75 |
| OFT-00477 | F | 1.25 | 1.25 | NA | NA | Healthy retina | Healthy retina | −7.5 | −0.75 | −7.875 | −8 | −1.5 | −8.75 |
| OFT-00517 | F | 1 | 0.8 | NA | NA | Tessellated fundus | Tessellated fundus | −6 | −1.25 | −6.625 | −5 | −2 | −6 |
| OFT-00529 * | F | NLP | 0.63 | NA | NA | Diffuse atrophy, Previous RD | Diffuse | NA | NA | NA | −0.25 | −1.75 | −1.25 |
| OFT-00623 | F | 0.5 | 0.67 | NA | NA | Tessellated fundus | Tessellated fundus | −6 | −2 | −7 | −2.75 | −0.75 | −3.5 |
| Family ID | First Diagnosis | Second Diagnosis | Gene | Transcript | Mutation | ACMG Criteria * | ACMG Result | Variant Type | Zygosity | Inheritance | Segregation Analysis Performed | De Novo /Inherited | Reported by |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFT-00074 | EoHM | NYSTAGMUS AND ESOTROPIA | TRPM1 | NM_002420.5 | Allele 1: c.3121C>T: p.Arg1041Trp Allele 2: c.1023+1G>A | PM2/PVS1, PM2, PP3, PP5 | VUS/P | Allele 1: Missense Allele 2: Splicing | Compound Hetero | AR | Yes | Allele 1: Maternal/Allele 2: Unknown | Allele 1: Novel/Allele 2: Miraldi Utz et al., 2018 [26] |
| OFT-00155 | EoHM | NYSTAGMUS | GPR143 | NM_000273.2 | c.1157G>A: p.Ser386Asn | PM2, PP1, PP2 | VUS | Missense | Hemi | X-linked | Yes | Maternal | Novel ** |
| CACNA1F | NM_005183.3 | c.2924G>A: p.Arg975Gln | PM2, PP1, PP3 | VUS | Missense | Hemi | X-linked | Yes | Maternal | Novel ** | |||
| OFT-00209 | EoHM | - | TIMP2 | NM_003255.5 | c.498C>G: p.Ile166Met | PM2, PP3 | VUS | Missense | Hetero | AD | Yes | Maternal | Novel ** |
| COL9A1 | NM_001851.6 | c.6G>T: p.Lys2Asn | PM2 | VUS | Missense | Hetero | AD | Yes | Unknown | Novel ** | |||
| OFT-00177 | EoHM | CONE-ROD DYSTROPHY AND SUBCAPSULAR CATARACT | CEP290 | NM_025114.4 | c.5777G>C: p.Arg1926Pro | PM2, PP3 | VUS | Missense | Hetero | S | No | Unknown | Wiszniewski et al., 2011 [27]; Sheck et al., 2018 [28]; Sallum et al., 2020 [29] |
| PCDH15 | NM_001142763.2 | c.5308_5313del: p.Ala1770_Pro1771del | PM2, PM4, PP3 | VUS | Deletion | Hetero | S | No | Unknown | Novel ** | |||
| OFT-00178 | EoHM | - | LRP5 | NM_002335.4 | c.4610C>T: p.Ala1537Val | PM2 | VUS | Missense | Hetero | AD | No | Paternal | Novel ** |
| OFT-00181 | EoHM | RETINAL DYSTROPHY | COL2A1 | NM_001844.5 | c.2818C>T: p.Arg940Ter | PVS1, PP5, PM2, PP3 | P | Nonsense | Hetero | AD | Yes | Maternal | Kondo et al., 2016 [30]; Maddirevula et al., 2018 [31]; Zhou et al., 2018 [4] |
| OFT-00223 | EoHM | - | PEX1 | NM_000466.3 | c.440T>C: p.Val147Ala | PM2, PP3 | VUS | Missense | Hetero | AD | Yes | Maternal | Novel ** |
| VDR | NM_001017536.2 | c.1223G>A: p.Arg408His | PM1, PM2 | VUS | Missense | Hetero | AD | No | Maternal | Novel ** | |||
| MMP9 | NM_004994.3 | c.822G>C: p.Glu274Asp | PM2 | VUS | Missense | Hetero | AD | Yes | Paternal | Novel ** | |||
| OFT-00092 | EoHM | RETINAL DYSTROPHY | KCNV2 | NM_133497.4 | c.458G>A: p.Arg153His | PM2, PP3 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| OFT-00097 | EoHM | NYSTAGMUS AND ASTIGMATISM | CFH | NM_000186.4 | c.907C>T: p.Arg303Trp | PM2, BP4 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| CACNA1F | NM_001256789.3 | c.4471C>T: p.Arg1491Ter | PVS1, PP5, PM2, PP3 | P | Nonsense | Hemi | AD | No | Maternal | Novel ** | |||
| OFT-00045 | EoHM | NYSTAGMUS AND RETINAL DYSTROPHY | PAX6 | NM_001258462.3 | c.262A>G: p.Ser88Gly | PS2, PM1, PM2, PP2, PP3 | P | Missense | Hetero | AD | Yes | De novo | Novel ** |
| OFT-00275 | EoHM | - | COL2A1 | NM_001844.5 | c.1783delC: p.Ala595LeufsTer34 | PVS1, PS2, PM2, PP3 | P | Frameshift | Hetero | AD | Yes | De novo | Novel ** |
| OFT-00332 | EoHM | - | ZNF644 | NM_201269.3 | c.1366A>T: p.Thr456Ser | PM2 | VUS | Missense | Hetero | AD | Yes | Maternal | Novel ** |
| CRYBB3 | NM_004076.5 | c.547G>T: p.Glu183 * | PM2, PP3 | VUS | Nonsense | Hetero | AD | Yes | Maternal | Novel ** | |||
| LRP5 | NM_002335.4 | c.263A>G: p.Lys88Arg | PM2 | VUS | Missense | Hetero | AD | Yes | Maternal | Novel ** | |||
| OFT-00343 | EoHM | - | OPA1 | NM_130837.3 | c.1294G>A: p.Val432Ile | PM1, PM2, PP2, PP3, PP5 | LP | Missense | Hetero | AD | Yes | Paternal | Stewart et al., 2008 [32]; Yu-Wai-Man et al., 2011 [33] |
| OFT-00191 | EoHM | - | COL11A1 | NM_001854.4 | c.2900G>T: p.Gly967Val | PM2, PP1, PP3 | LP | Missense | Hetero | AD | Yes | Paternal | Novel ** |
| OFT-00391 | EoHM | ASTIGMATISM | CRYGC | NM_020989.4 | c.179G>A: p.Arg60Gln | PM2, PP2 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| RDH5 | NM_001199771.2 | c.683G>A: p.Arg228Gln | PM2, PP2, BP4 | VUS | Missense | Hetero | S | No | Unknown | Novel ** | |||
| OFT-00407 | EoHM | CONE-ROD DYSTROPHY | ARL6 | NM_177976.3 | c.362G>A: p.Arg121His | PM2, PM3, PP2, PP3, PP5 | LP | Missense | Homo | AR | Yes | Maternal and Paternal | Patel et al., 2016 [34]; Abouelhoda et al., 2016 [35] |
| OFT-00429 | EoHM | - | MMP9 | NM_004994.3 | c.1270C>A: p.Arg424Ser | PM2, BP4 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| IGF1R | NM_000875.5 | c.3784A>C: p.Ile1262Leu | PM2, PP3 | VUS | Missense | Hetero | S | No | Unknown | Novel ** | |||
| OFT-00436 | EoHM | - | MMP10 | NM_002425.3 | c.497-2A>G | PP3, BS1 | VUS | Splicing | Hetero | AD | Yes | Maternal | Novel ** |
| OFT-00453 | EoHM | RETINAL DYSTROPHY AND PERSISTENT FETAL VASCULATURE RIGHT EYE | COL2A1 | NM_001844.5 | c.157C>T: p.Arg53Trp | PM2, PP2, PP3 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| TRPM1 | NM_001252020.1 | c.3618C>G: p.Phe1206Leu | PM2 | VUS | Missense | Hetero | S | No | Unknown | Novel ** | |||
| OFT-00463 | EoHM | - | EPHA2 | NM_004431.5 | c.308G>A: p.Arg103His | PM2, PP3 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| OFT-00474 | EoHM | - | MERTK | NM_006343.3 | c.2264G>A: p.Arg755His | PM2, PP3 | VUS | Missense | Hetero | AR | Yes | Maternal | Novel ** |
| OFT-00490 | EoHM | - | COL11A1 | NM_001854.4 | c.1021G>C: p.Glu341Gln | PM2 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| TRPM1 | NM_001252020.1 | c.4550C>T: p.Thr1517Met | PM2, BP4 | VUS | Missense | Hetero | S | No | Unknown | Novel ** | |||
| OFT-00493 | EoHM | - | CRYGA | NM_014617.4 | c.287A>G: p.Asp96Gly | PM2, BP4 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| OFT-00506 | EoHM | - | ZNRF3 | NM_001206998.2 | c.2221G>A: p.Glu741Lys | PM2, BP4 | VUS | Missense | Hetero | AD | Yes | Unknown | Novel ** |
| OFT-00533 | EoHM | - | SCO2 | NM_001169111.1 | c.334C>T: p.Arg112Trp | PM2, PP5 | VUS | Missense | Hetero | S | No | Unknown | Jiang et al., 2015 [36] |
| OFT-00546 | EoHM | - | LAMA2 | NM_000426.4 | c.6880G>T: p.Val2294Leu | PM2, PP3 | VUS | Missense | Hetero | AR | Yes | Paternal | Novel ** |
| OFT-00554 | EoHM | - | SCO2 | NM_001169111.1 | c.341G>A: p.Arg114His | PS3, PM2, PP3, PP5 | LP | Missense | Hetero | AD | Yes | Maternal or Paternal | Tran-Viet et al., 2013 [37]; Pacheu-Grau et al., 2015 [38]; Kars et al., 2021 [39] |
| OFT-00559 | EoHM | NYSTAGMUS | NDP | NM_000266.4 | c.313_314delGCinsTT: p.Ala105Leu | PM1, PM2, PM5, PS1, PP2, PP3 | P | Deletion/Insertion | Hemi | S | No | Unknown | Novel ** |
| OFT-00568 | EoHM | - | PEX1 | NM_000466.3 | c.3250A>G: p.Met1084Val | PM2 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
| OFT-00586 | EoHM | RETINAL DYSTROPHY LEFT EYE | MMP1 | NM_002421.4 | c.1389G>A: p.Trp463Ter | PP3, BS1 | VUS | Nonsense | Hetero | S | No | Unknown | Novel ** |
| OFT-00590 | EoHM | - | COL11A1 | NM_001854.4 | c.1570C>T: p.Arg524Trp | PM2, PP3 | LP | Missense | Hetero | S | No | Unknown | Novel ** |
| OFT-00601 | EoHM | - | GPR143 | NM_000273.3 | c.47C>A: p.Ala16Glu | PM2, PP2, PP3, BP6 | VUS | Missense | Hemi | X-linked | Yes | Maternal | Novel ** |
| OFT-00630 | EoHM | - | CRYBA1 | NM_005208.5 | c.190C>T: p.Arg64Trp | PM2, PP3 | VUS | Missense | Hetero | S | No | Unknown | Novel ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Iglesias, E.; López-Vázquez, A.; Noval, S.; Nieves-Moreno, M.; Granados-Fernández, M.; Arruti, N.; Rosa-Pérez, I.; Pacio-Míguez, M.; Montaño, V.E.F.; Rodríguez-Solana, P.; et al. Next-Generation Sequencing Screening of 43 Families with Non-Syndromic Early-Onset High Myopia: A Clinical and Genetic Study. Int. J. Mol. Sci. 2022, 23, 4233. https://doi.org/10.3390/ijms23084233
González-Iglesias E, López-Vázquez A, Noval S, Nieves-Moreno M, Granados-Fernández M, Arruti N, Rosa-Pérez I, Pacio-Míguez M, Montaño VEF, Rodríguez-Solana P, et al. Next-Generation Sequencing Screening of 43 Families with Non-Syndromic Early-Onset High Myopia: A Clinical and Genetic Study. International Journal of Molecular Sciences. 2022; 23(8):4233. https://doi.org/10.3390/ijms23084233
Chicago/Turabian StyleGonzález-Iglesias, Eva, Ana López-Vázquez, Susana Noval, María Nieves-Moreno, María Granados-Fernández, Natalia Arruti, Irene Rosa-Pérez, Marta Pacio-Míguez, Victoria E. F. Montaño, Patricia Rodríguez-Solana, and et al. 2022. "Next-Generation Sequencing Screening of 43 Families with Non-Syndromic Early-Onset High Myopia: A Clinical and Genetic Study" International Journal of Molecular Sciences 23, no. 8: 4233. https://doi.org/10.3390/ijms23084233
APA StyleGonzález-Iglesias, E., López-Vázquez, A., Noval, S., Nieves-Moreno, M., Granados-Fernández, M., Arruti, N., Rosa-Pérez, I., Pacio-Míguez, M., Montaño, V. E. F., Rodríguez-Solana, P., del Pozo, A., Santos-Simarro, F., & Vallespín, E. (2022). Next-Generation Sequencing Screening of 43 Families with Non-Syndromic Early-Onset High Myopia: A Clinical and Genetic Study. International Journal of Molecular Sciences, 23(8), 4233. https://doi.org/10.3390/ijms23084233