
Dissecting the Molecular Mechanisms Surrounding Post-COVID-19 Syndrome and Neurological Features

Abstract
:1. Introduction
2. Post-COVID-19 Syndrome Characterization
2.1. Previous SARS Outbreaks and Long-Term Complications
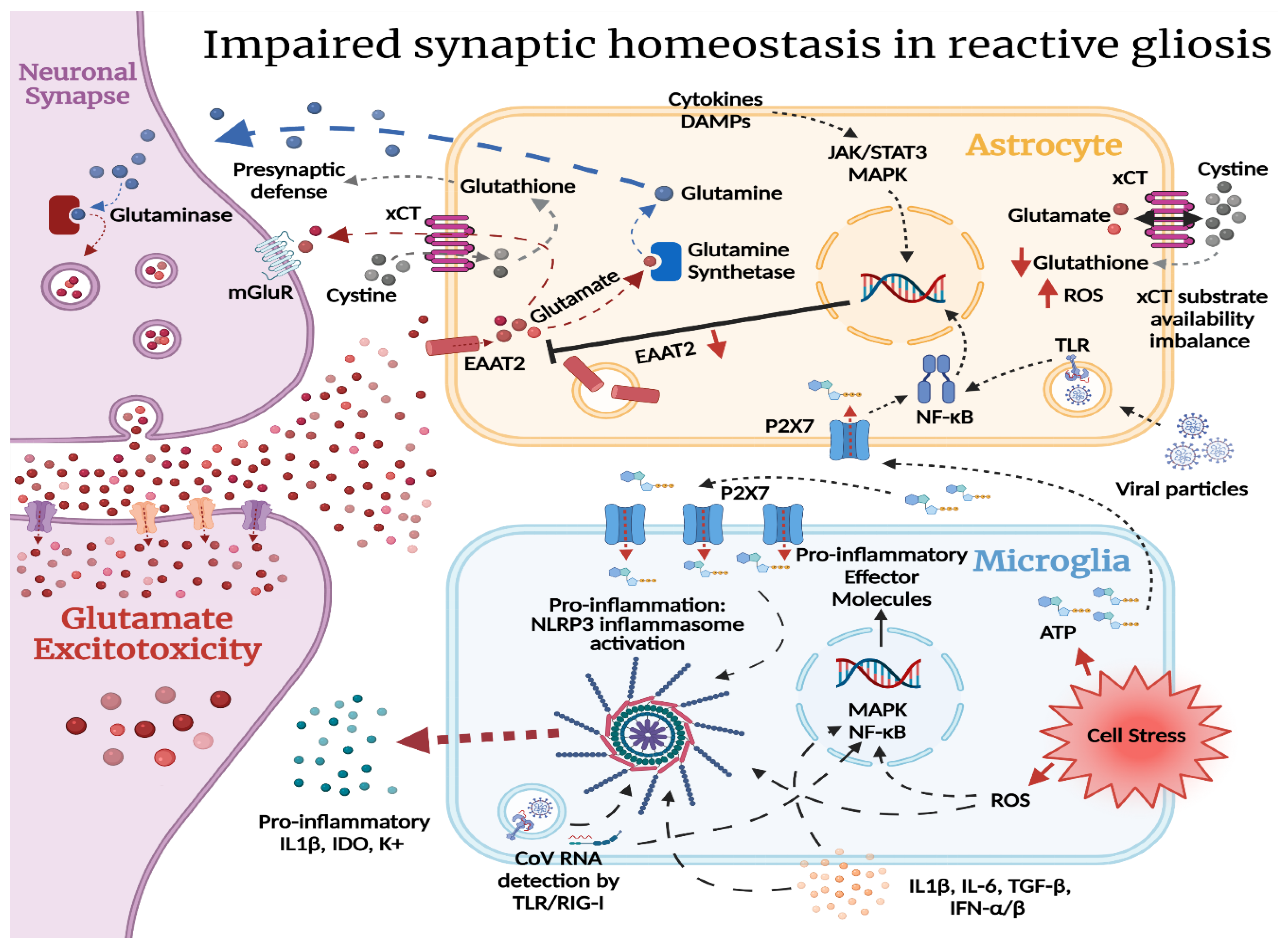
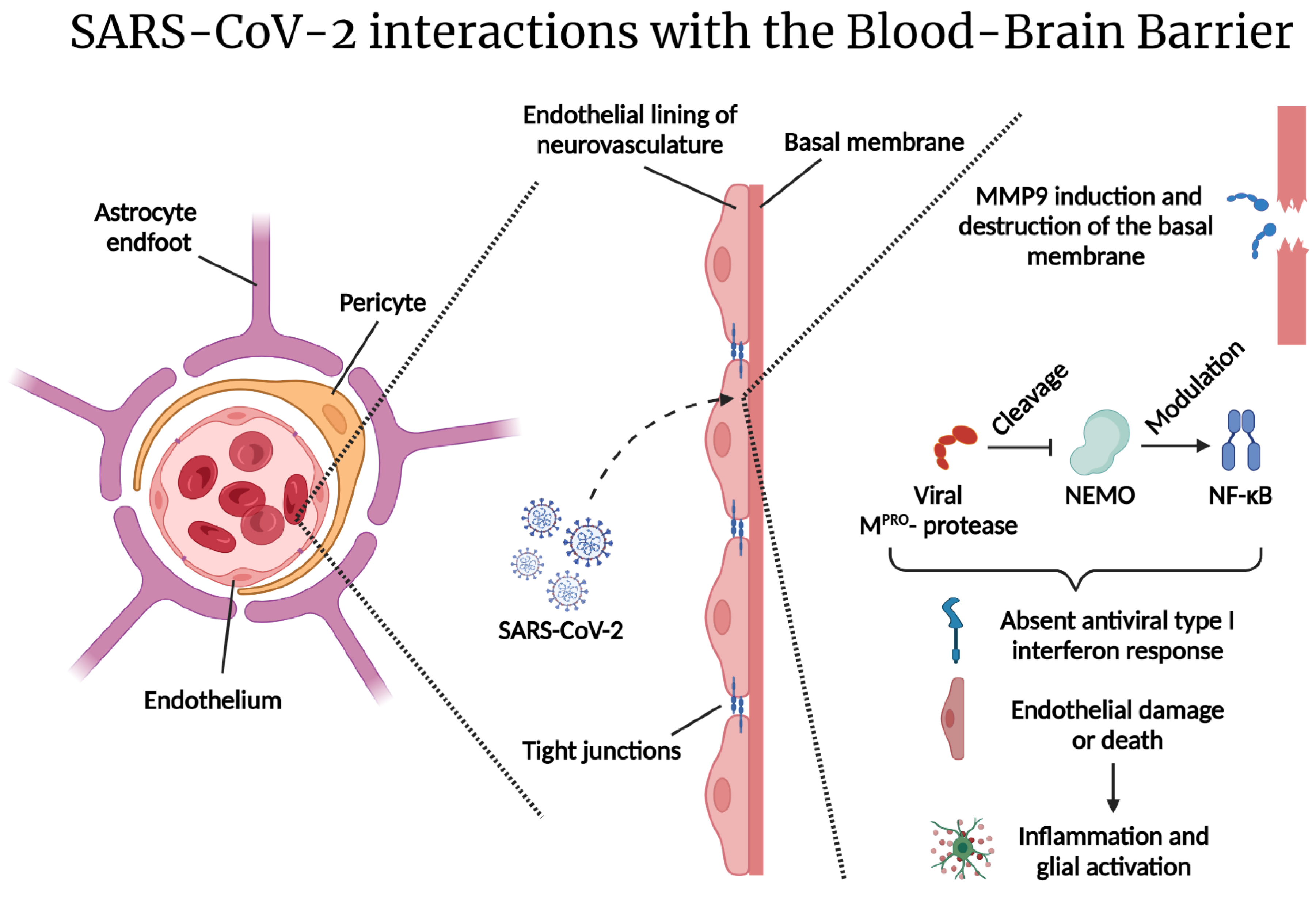
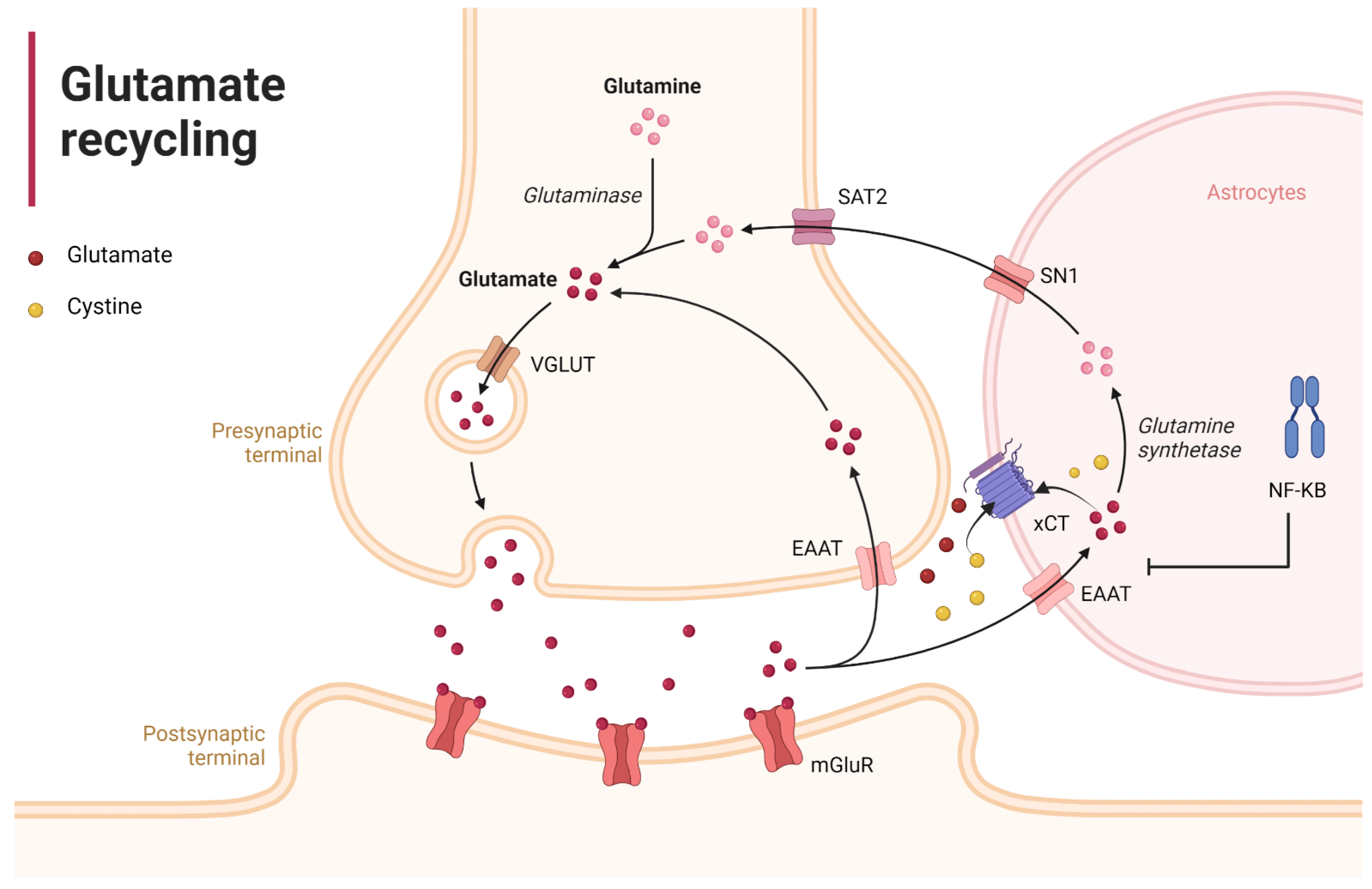
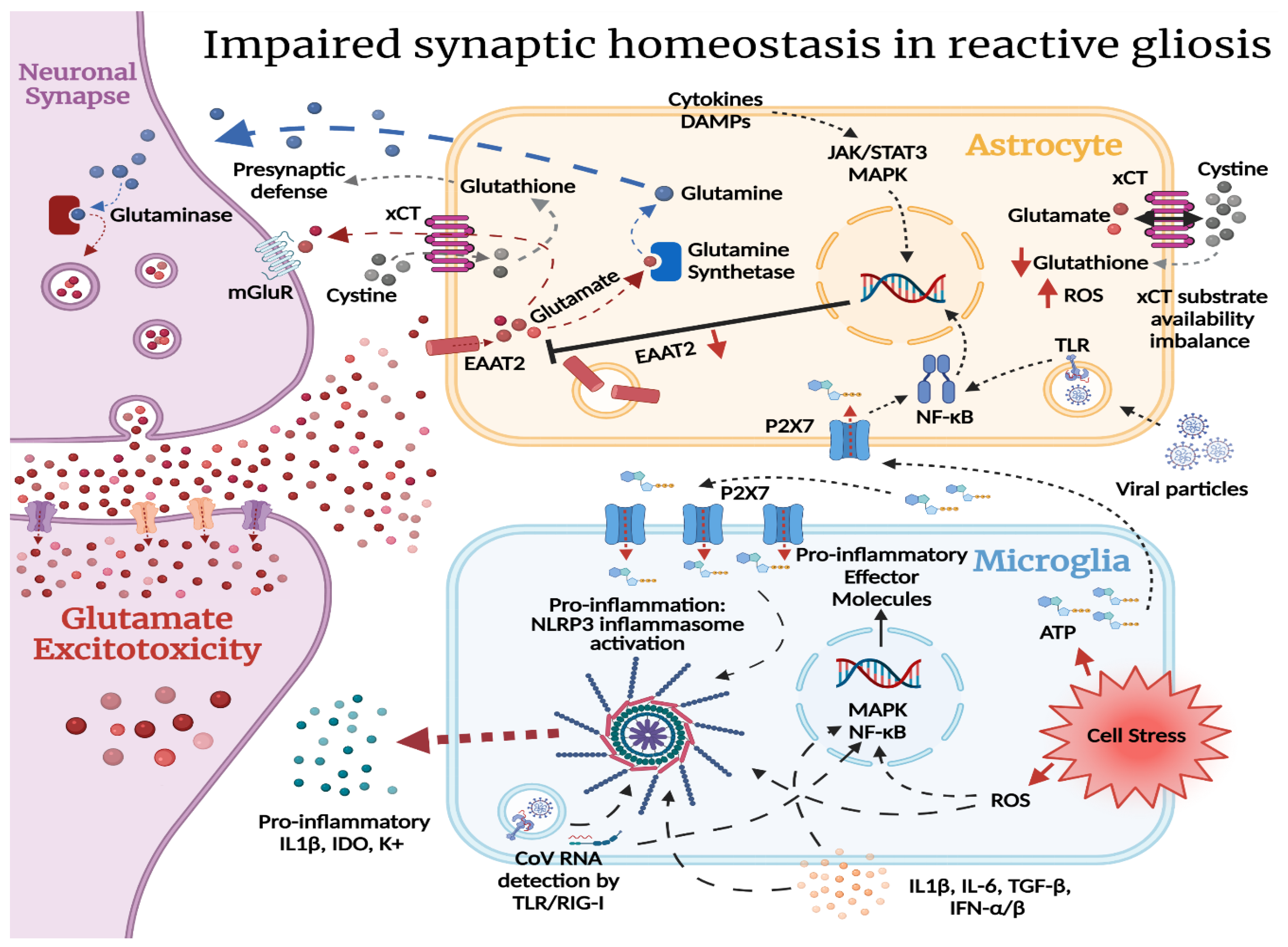
2.2. Reactive Gliosis and Excitotoxicity in the Wake of SARS-CoV-2 CNS Injury
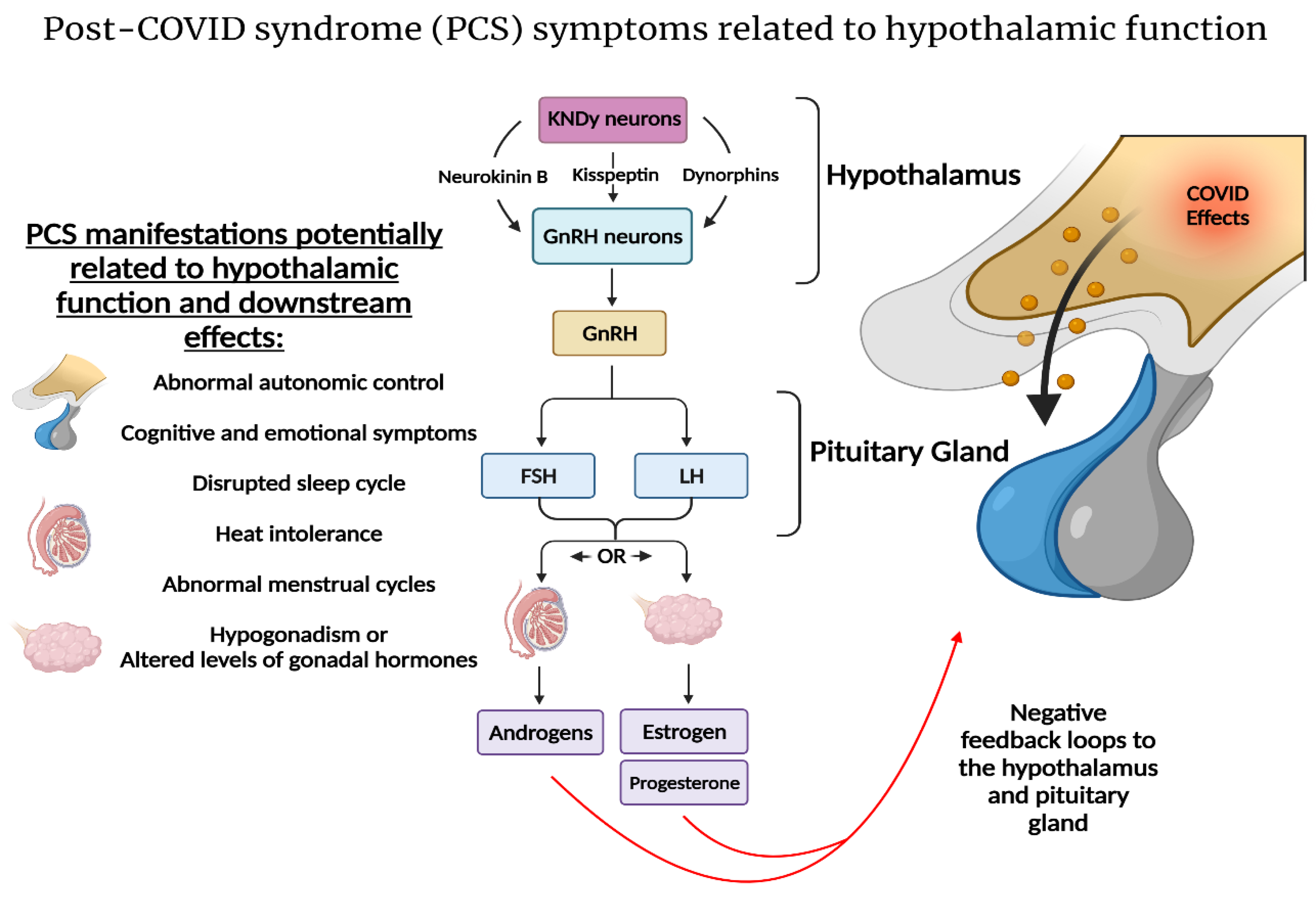
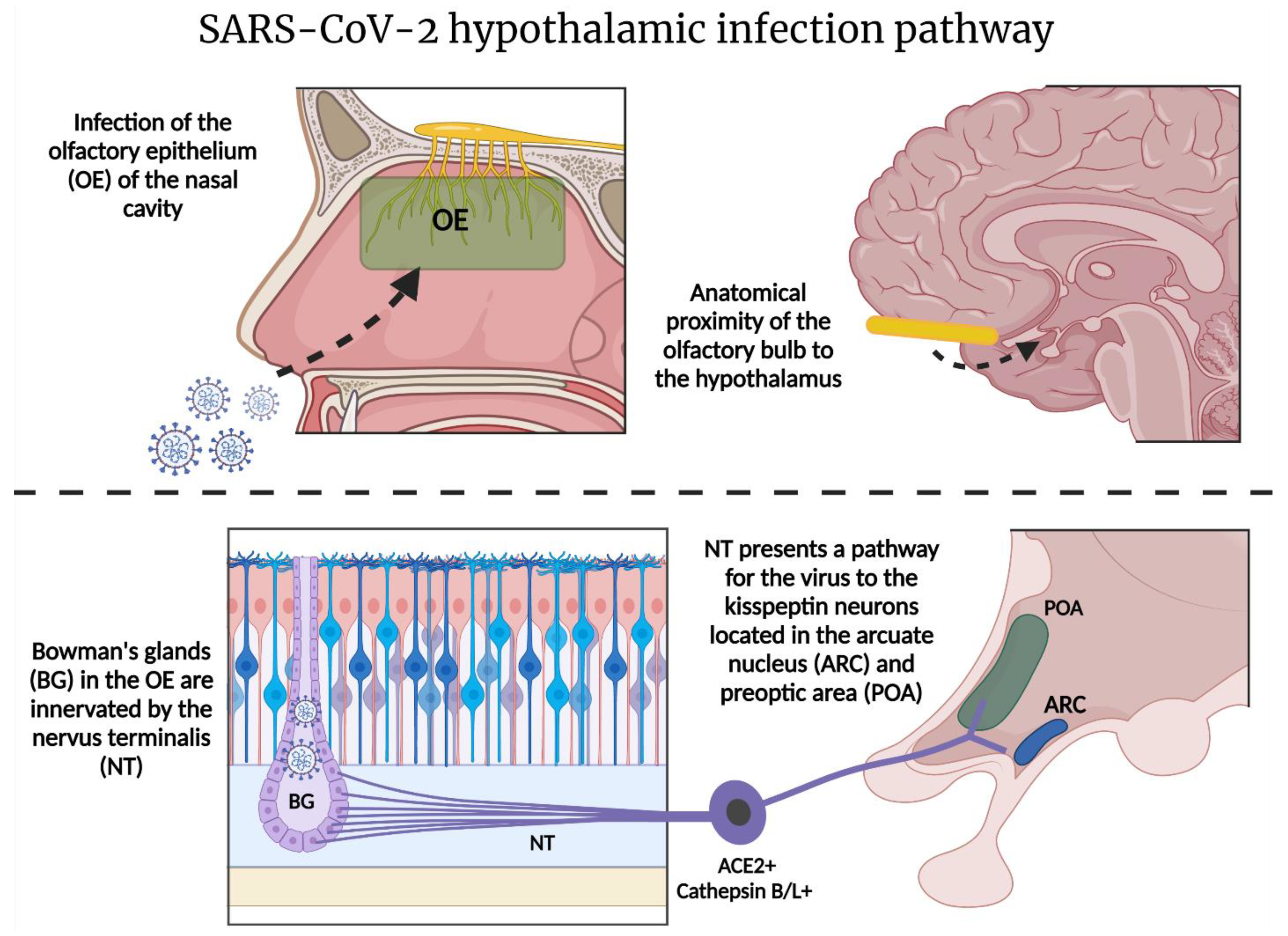
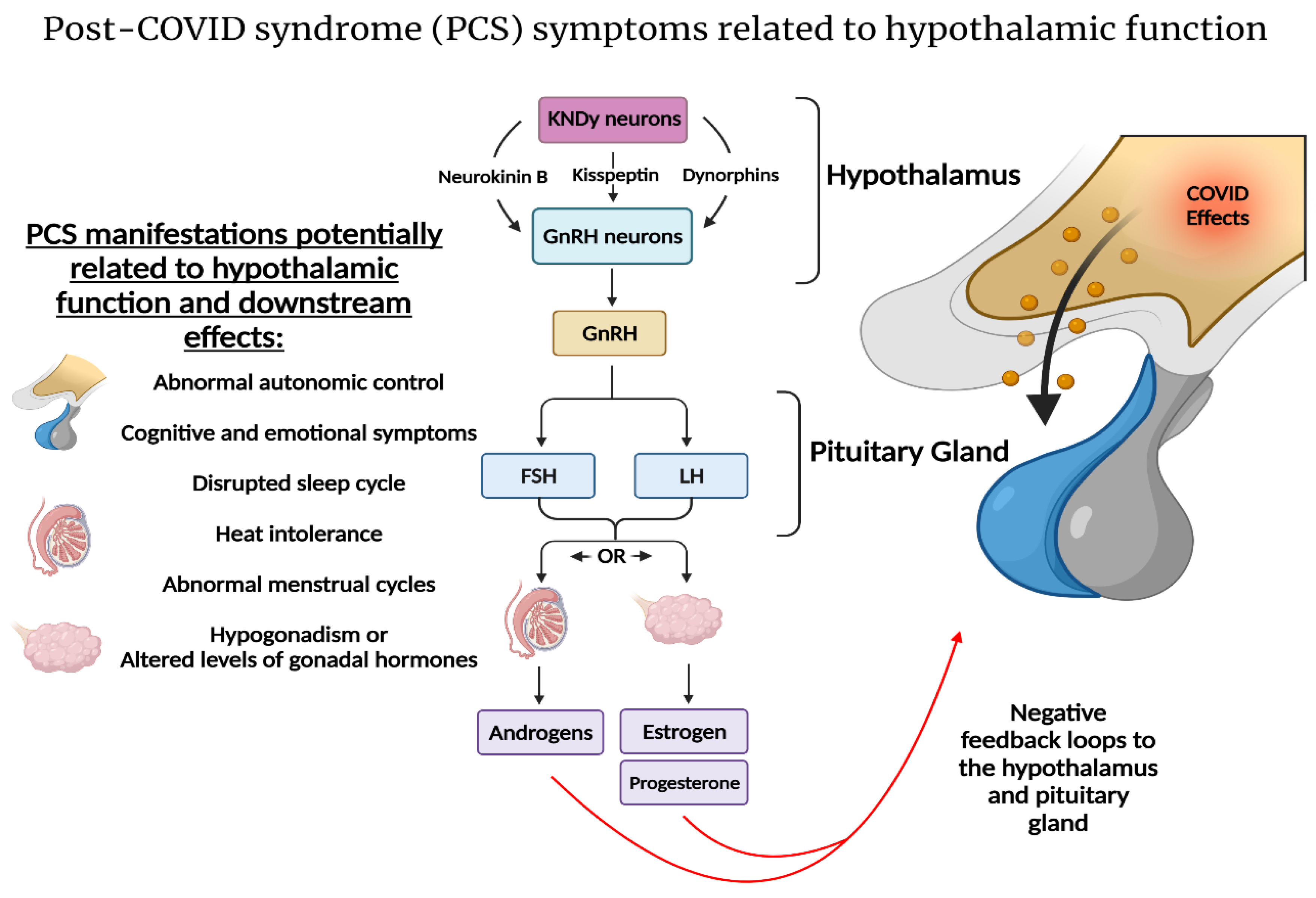
2.3. SARS-CoV-2 and the Hypothalamic–Kisspeptin Neurons
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Baig, A.M. Chronic COVID syndrome: Need for an appropriate medical terminology for long-COVID and COVID long-haulers. J. Med. Virol. 2021, 93, 2555–2556. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [Google Scholar] [CrossRef]
- Rubin, R. As Their Numbers Grow, COVID-19 “Long Haulers” Stump Experts. JAMA 2020, 324, 1381–1383. [Google Scholar] [CrossRef]
- Datta, S.D.; Talwar, A.; Lee, J.T. A Proposed Framework and Timeline of the Spectrum of Disease Due to SARS-CoV-2 Infection: Illness Beyond Acute Infection and Public Health Implications. JAMA 2020, 324, 2251–2252. [Google Scholar] [CrossRef]
- Post-COVID Conditions: Information for Healthcare Providers [Internet]. Available online: https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-care/post-covid-conditions.html (accessed on 21 July 2021).
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Huang, L.; Yao, Q.; Gu, X.; Wang, Q.; Ren, L.; Wang, Y.; Hu, P.; Guo, L.; Liu, M.; Xu, J.; et al. 1-year outcomes in hospital survivors with COVID-19: A longitudinal cohort study. Lancet 2021, 398, 747–758. [Google Scholar] [CrossRef]
- Sigfrid, L.; Drake, T.M.; Pauley, E.; Jesudason, E.C.; Olliaro, P.; Lim, W.S.; Gillesen, A.; Berry, C.; Lowe, D.J.; McPeake, J.; et al. Long COVID in adults discharged from UK hospitals after COVID-19: A prospective, multicentre cohort study using the ISARIC WHO Clinical Characterisation Protocol. Lancet Reg. Health Eur. 2021, 8, 100186. [Google Scholar] [CrossRef]
- Whitaker, M.; Elliott, J.; Chadeau-Hyam, M.; Riley, S.; Darzi, A.; Cooke, G.; Ward, H.; Elliott, P. Persistent symptoms following SARS-CoV-2 infection in a random community sample of 508,707 people. medRxiv 2021. [Google Scholar] [CrossRef]
- Blomberg, B.; Mohn, K.G.-I.; Brokstad, K.A.; Zhou, F.; Linchausen, D.W.; Hansen, B.-A.; Lartey, S.; Onyango, T.B.; Kuwelker, K.; Sævik, M.; et al. Long COVID in a prospective cohort of home-isolated patients. Nat. Med. 2021, 27, 1607–1613. [Google Scholar] [CrossRef]
- Torjesen, I. COVID-19: Middle aged women face greater risk of debilitating long term symptoms. BMJ 2021, 372, n829. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Moulin, T.C.; Schiöth, H.B. Sex differences in COVID-19: The role of androgens in disease severity and progression. Endocrine 2020, 71, 3–8. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895. [Google Scholar] [CrossRef]
- Moreno-Pérez, O.; Merino, E.; Leon-Ramirez, J.-M.; Andres, M.; Ramos, J.M.; Arenas-Jiménez, J.; Asensio, S.; Sanchez, R.; Ruiz-Torregrosa, P.; Galan, I.; et al. Post-acute COVID-19 syndrome. Incidence and risk factors: A Mediterranean cohort study. J. Infect. 2021, 82, 378–383. [Google Scholar] [CrossRef]
- Iqbal, F.M.; Lam, K.; Sounderajah, V.; Clarke, J.M.; Ashrafian, H.; Darzi, A. Characteristics and predictors of acute and chronic post-COVID syndrome: A systematic review and meta-analysis. eClinicalMedicine 2021, 36, 100899. [Google Scholar] [CrossRef]
- Davis, H.E.; Assaf, G.S.; McCorkell, L.; Wei, H.; Low, R.J.; Re’Em, Y.; Redfield, S.; Austin, J.P.; Akrami, A. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. eClinicalMedicine 2021, 38, 101019. [Google Scholar] [CrossRef]
- Zhao, Y.M.; Shang, Y.M.; Song, W.B.; Li, Q.Q.; Xie, H.; Xu, Q.F.; Jia, J.L.; Li, L.M.; Mao, H.L.; Zhou, X.M.; et al. Follow-up study of the pulmonary function and related physiological characteristics of COVID-19 survivors three months after recovery. EClinicalMedicine 2020, 25, 100463. [Google Scholar] [CrossRef]
- Torres-Castro, R.; Vasconcello-Castillo, L.; Alsina-Restoy, X.; Solis-Navarro, L.; Burgos, F.; Puppo, H.; Vilaró, J. Respiratory function in patients post-infection by COVID-19: A systematic review and meta-analysis. Pulmonology 2020, 27, 328–337. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F.; Group for the GAC-19 P-ACS. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Halpin, S.J.; McIvor, C.; Whyatt, G.; Adams, A.; Harvey, O.; McLean, L.; Walshaw, C.; Kemp, S.; Corrado, J.; Singh, R.; et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: A cross-sectional evaluation. J. Med. Virol. 2021, 93, 1013–1022. [Google Scholar] [CrossRef]
- Kamal, M.; Omirah, M.A.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pract. 2021, 75, e13746. [Google Scholar] [CrossRef]
- Seeßle, J.; Waterboer, T.; Hippchen, T.; Simon, J.; Kirchner, M.; Lim, A.; Müller, B.; Merle, U. Persistent symptoms in adult patients one year after COVID-19: A prospective cohort study. Clin. Infect. Dis. 2021, 1–8. [Google Scholar]
- Townsend, L.; Dyer, A.H.; Jones, K.; Dunne, J.; Mooney, A.; Gaffney, F.; O’Connor, L.; Leavy, D.; O’Brien, K.; Dowds, J.; et al. Persistent fatigue following SARS-CoV-2 infection is common and independent of severity of initial infection. PLoS ONE 2020, 15, e0240784. [Google Scholar] [CrossRef]
- Ramakrishnan, R.K.; Kashour, T.; Hamid, Q.; Halwani, R.; Tleyjeh, I.M. Unraveling the Mystery Surrounding Post-Acute Sequelae of COVID-19. Front. Immunol. 2021, 12, 2574. [Google Scholar] [CrossRef]
- Afrin, L.B.; Weinstock, L.B.; Molderings, G.J. COVID-19 hyperinflammation and post-COVID-19 illness may be rooted in mast cell activation syndrome. Int. J. Infect. Dis. 2020, 100, 327–332. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deitchman, A.N.; Torres, L.; Iyer, N.S.; Munter, S.E.; Nixon, C.C.; Donatelli, J.; Thanh, C.; Takahashi, S.; Hakim, J.; et al. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 2021, 36, 109518. [Google Scholar] [CrossRef]
- Hui, D.S.; Joynt, G.M.; Wong, K.T.; Gomersall, C.D.; Li, T.S.; Antonio, G.; Ko, F.W.; Chan, M.C.; Chan, D.P.; Tong, M.W.; et al. Impact of severe acute respiratory syndrome [SARS] on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax 2005, 60, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.S.; Wong, K.T.; Ko, F.W.S.; Tam, L.-S.; Chan, D.P.; Woo, J.; Sung, J.J.Y. The 1-Year Impact of Severe Acute Respiratory Syndrome on Pulmonary Function, Exercise Capacity, and Quality of Life in a Cohort of Survivors. Chest 2005, 128, 2247–2261. [Google Scholar] [CrossRef] [Green Version]
- Das, K.M.; Lee, E.Y.; Singh, R.; Enani, M.A.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J. Radiol. Imaging 2021, 27, 342–349. [Google Scholar] [CrossRef]
- Park, W.B.; Jun, K.I.; Kim, G.; Choi, J.-P.; Rhee, J.-Y.; Cheon, S.; Lee, C.H.; Park, J.-S.; Kim, Y.; Joh, J.-S.; et al. Correlation between Pneumonia Severity and Pulmonary Complications in Middle East Respiratory Syndrome. J. Korean Med. Sci. 2018, 33, e169. [Google Scholar] [CrossRef]
- Ngai, J.C.; Ko, F.W.; Ng, S.S.; TO, K.W.; Tong, M.; Hui, D.S. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology 2010, 15, 543. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.H.B.; Wing, Y.K.; Yu, M.W.M.; Leung, C.M.; Ma, R.C.; Kong, A.P.; So, W.Y.; Fong, S.Y.Y.; Lam, S.P. Mental Morbidities and Chronic Fatigue in Severe Acute Respiratory Syndrome Survivors: Long-term Follow-up. Arch. Intern. Med. 2009, 169, 2142–2147. [Google Scholar] [CrossRef] [Green Version]
- Moldofsky, H.; Patcai, J. Chronic widespread musculoskeletal pain, fatigue, depression and disordered sleep in chronic post-SARS syndrome; a case-controlled study. BMC Neurol. 2011, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Shin, H.S.; Park, H.Y.; Kim, J.L.; Lee, J.J.; Lee, H.; Won, S.D.; Han, W. Depression as a Mediator of Chronic Fatigue and Post-Traumatic Stress Symptoms in Middle East Respiratory Syndrome Survivors. Psychiatry Investig. 2019, 16, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, K.; Skoog, E.; Jones, V.; Sandelin, L.L.; Björling, C.; Fridenström, E.; Edvinsson, M.; Mårtensson, A.; Olsen, B. A comprehensive clinical and laboratory evaluation of 224 patients with persistent symptoms attributed to presumed tick-bite exposure. PLoS ONE 2021, 16, e0247384. [Google Scholar] [CrossRef]
- Comella, P.H.; Gonzalez-Kozlova, E.; Kosoy, R.; Charney, A.; Peradejordi, I.; Chandrasekar, S.; Tyler, S.; Wang, W.; Losic, B.; Zhu, J.; et al. A Molecular network approach reveals shared cellular and molecular signatures between chronic fatigue syndrome and other fatiguing illnesses. medRxiv 2021. [Google Scholar] [CrossRef]
- Chou, S.H.Y.; Beghi, E.; Helbok, R.; Moro, E.; Sampson, J.; Altamirano, V.; Mainali, S.; Bassetti, C.; Suarez, J.I.; McNett, M.; et al. Global Incidence of Neurological Manifestations Among Patients Hospitalized With COVID-19—A Report for the GCS-NeuroCOVID Consortium and the ENERGY Consortium. JAMA Netw. Open 2021, 4, e2112131. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Johansson, A.; Mohamed, M.S.; Moulin, T.C.; Schiöth, H.B. Neurological manifestations of COVID-19: A comprehensive literature review and discussion of mechanisms. J. Neuroimmunol. 2021, 358, 577658. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, Ü.; et al. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, L.; Bao, L.; Liu, J.; Zhu, H.; Lv, Q.; Liu, R.; Chen, W.; Tong, W.; Wei, Q.; et al. SARS-CoV-2 crosses the blood–brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct. Target. Ther. 2021, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2020, 24, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bilinska, K.; von Bartheld, C.S.; Butowt, R. Expression of the ACE2 Virus Entry Protein in the Nervus Terminalis Reveals the Potential for an Alternative Route to Brain Infection in COVID-19. Front. Cell. Neurosci. 2021, 15, 674123. [Google Scholar] [CrossRef]
- Frank, M.G.; Nguyen, K.H.; Ball, J.B.; Hopkins, S.; Kelley, T.; Baratta, M.V.; Fleshner, M.; Maier, S.F. SARS-CoV-2 spike S1 subunit induces neuroinflammatory, microglial and behavioral sickness responses: Evidence of PAMP-like properties. Brain Behav. Immun. 2022, 100, 267–277. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Vendramini, P.H.; Valença, A.G.F.; Antunes, A.S.L.M.; Brandão-Teles, C.; da Silva Zuccoli, G.; Reis-de-Oliveira, G.; Silva-Costa, L.C.; et al. SARS-CoV-2 infects brain astrocytes of COVID-19 patients and impairs neuronal viability. medRxiv 2020, 16. [Google Scholar] [CrossRef]
- Virhammar, J.; Nääs, A.; Fällmar, D.; Cunningham, J.L.; Klang, A.; Ashton, N.J.; Jackmann, S.; Westman, G.; Frithiof, R.; Blennow, K.; et al. Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur. J. Neurol. 2020, 28, 3324–3331. [Google Scholar] [CrossRef]
- Kanberg, N.; Ashton, N.J.; Andersson, L.-M.; Yilmaz, A.; Lindh, M.; Nilsson, S.; Price, R.W.; Blennow, K.; Zetterberg, H.; Gisslén, M. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology 2020, 95, e1754–e1759. [Google Scholar] [CrossRef]
- Lee, M.-H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with COVID-19. N. Engl. J. Med. 2021, 384, 481–483. [Google Scholar] [CrossRef]
- Tavčar, P.; Potokar, M.; Kolenc, M.; Korva, M.; Avšič-Županc, T.; Zorec, R.; Jorgačevski, J. Neurotropic Viruses, Astrocytes, and COVID-19. Front. Cell. Neurosci. 2021, 15, 123. [Google Scholar] [CrossRef]
- Schousboe, A. Metabolic signaling in the brain and the role of astrocytes in control of glutamate and GABA neurotransmission. Neurosci. Lett. 2019, 689, 11–13. [Google Scholar] [CrossRef] [PubMed]
- De Pittà, M.; Brunel, N.; Volterra, A. Astrocytes: Orchestrating synaptic plasticity? Neuroscience 2016, 323, 43–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Lee, S.-G.; Kegelman, T.P.; Su, Z.-Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of Excitatory Amino Acid Transporter-2 (EAAT2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell. Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A Local Glutamate-Glutamine Cycle Sustains Synaptic Excitatory Transmitter Release. Neuron 2014, 81, 888–900. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef]
- Moran, M.M.; McFarland, K.; Melendez, R.I.; Kalivas, P.W.; Seamans, J.K. Cystine/Glutamate Exchange Regulates Metabotropic Glutamate Receptor Presynaptic Inhibition of Excitatory Transmission and Vulnerability to Cocaine Seeking. J. Neurosci. 2005, 25, 6389–6393. [Google Scholar] [CrossRef] [Green Version]
- Saggu, R.; Schumacher, T.; Gerich, F.; Rakers, C.; Tai, K.; Delekate, A.; Petzold, G.C. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol. Commun. 2016, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Parpura, V. Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol. Dis. 2016, 85, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Matott, M.P.; Kline, D.D.; Hasser, E.M. Glial EAAT2 regulation of extracellular nTS glutamate critically controls neuronal activity and cardiorespiratory reflexes. J. Physiol. 2017, 595, 6045–6063. [Google Scholar] [CrossRef]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.-M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2014, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal, A.; Vidos, C.; Monteverde Busso, M.; Cieri, M.B.; Ramos, A.J. Pathological Neuroinflammatory Conversion of Reactive Astrocytes Is Induced by Microglia and Involves Chromatin Remodeling. Front. Pharmacol. 2021, 12, 1448. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.E.; Oliveira-Giacomelli, Á.; Glaser, T.; Arnaud-Sampaio, V.F.; Andrejew, R.; Dieckmann, L.; Baranova, J.; Lameu, C.; Ratajczak, M.Z.; Ulrich, H. Hyperactivation of P2X7 receptors as a culprit of COVID-19 neuropathology. Mol. Psychiatry 2020, 26, 1044–1059. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- John, G.R.; Simpson, J.E.; Woodroofe, M.N.; Lee, S.C.; Brosnan, C.F. Extracellular nucleotides differentially regulate interleukin-1beta signaling in primary human astrocytes: Implications for inflammatory gene expression. J. Neurosci. 2001, 21, 4134–4142. [Google Scholar] [CrossRef]
- Roman, M.; Irwin, M.R. Novel neuroimmunologic therapeutics in depression: A clinical perspective on what we know so far. Brain Behav. Immun. 2020, 83, 7–21. [Google Scholar] [CrossRef]
- Kedor, C.; Freitag, H.; Meyer-Arndt, L.; Wittke, K.; Steinbeis, F.; Haffke, M.; Gordon, R.; Heidecker, B.; Volk, H.D.; Skurk, C.; et al. Chronic COVID-19 Syndrome and Chronic Fatigue Syndrome [ME/CFS] following the first pandemic wave in Germany—A first analysis of a prospective observational study. medRxiv 2021. [Google Scholar] [CrossRef]
- Noda, M.; Ifuku, M.; Hossain, M.S.; Katafuchi, T. Glial Activation and Expression of the Serotonin Transporter in Chronic Fatigue Syndrome. Front. Psychiatry 2018, 9, 589. [Google Scholar] [CrossRef]
- Barnden, L.R.; Crouch, B.; Kwiatek, R.; Burnet, R.; Mernone, A.; Chryssidis, S.; Scroop, G.; Del Fante, P. A brain MRI study of chronic fatigue syndrome: Evidence of brainstem dysfunction and altered homeostasis. NMR Biomed. 2011, 24, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Nijs, J.; George, S.Z.; Clauw, D.J.; Fernández-De-Las-Peñas, C.; Kosek, E.; Ickmans, K.; Fernández-Carnero, J.; Polli, A.; Kapreli, E.; Huysmans, E.; et al. Central sensitisation in chronic pain conditions: Latest discoveries and their potential for precision medicine. Lancet Rheumatol. 2021, 3, e383–e392. [Google Scholar] [CrossRef]
- Druce, K.L.; McBeth, J. Central sensitization predicts greater fatigue independently of musculoskeletal pain. Rheumatology 2019, 58, 1923–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeus, M.; Nijs, J. Central sensitization: A biopsychosocial explanation for chronic widespread pain in patients with fibromyalgia and chronic fatigue syndrome. Clin. Rheumatol. 2006, 26, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijs, J.; Meeus, M.; Van Oosterwijck, J.; Ickmans, K.; Moorkens, G.; Hans, G.; De Clerck, L.S. In the mind or in the brain? Scientific evidence for central sensitisation in chronic fatigue syndrome. Eur. J. Clin. Investig. 2012, 42, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liang, J.; Wang, J.; Fei, Z.; Qin, G.; Zhang, D.; Zhou, J.; Chen, L. Up-regulation of astrocyte excitatory amino acid transporter 2 alleviates central sensitization in a rat model of chronic migraine. J. Neurochem. 2020, 155, 370–389. [Google Scholar] [CrossRef]
- Tang, J.; Bair, M.; Descalzi, G. Reactive Astrocytes: Critical Players in the Development of Chronic Pain. Front. Psychiatry 2021, 12, 809. [Google Scholar] [CrossRef]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. S1), S10–S28. [Google Scholar] [CrossRef]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.H.; Wood, J.; Yalcin, B.; Taylor, K.R.; Dutton, S.; Acosta-Alvarez, L.; et al. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kanberg, N.; Simrén, J.; Edén, A.; Andersson, L.-M.; Nilsson, S.; Ashton, N.J.; Sundvall, P.-D.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Neurochemical signs of astrocytic and neuronal injury in acute COVID-19 normalizes during long-term follow-up. eBioMedicine 2021, 70, 103512. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Butowt, R.; Meunier, N.; Bryche, B.; von Bartheld, C.S. The olfactory nerve is not a likely route to brain infection in COVID-19: A critical review of data from humans and animal models. Acta Neuropathol. 2021, 141, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. {SARS}-{CoV}-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Sonne, J.; Lopez-Ojeda, W. Neuroanatomy, Cranial Nerve; StatPearls: Treasure Island, FL, USA, 19 November 2020. [Google Scholar]
- Pineda, A.G.; Leon-Sarmiento, F.E.; Doty, R.L. Cranial nerve 13. Handb. Clin. Neurol. 2019, 164, 135–144. [Google Scholar]
- Nampoothiri, S.; Sauve, F.; Ternier, G.; Fernandois, D.; Coelho, C.; Imbernon, M.; Deligia, E.; Perbet, R.; Florent, V.; Baroncini, M.; et al. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Schwanzel-Fukuda, M.; Morrell, J.I.; Pfaff, D.W. Ontogenesis of neurons producing luteinizing hormone-releasing hormone (LHRH) in the nervus terminalis of the rat. J. Comp. Neurol. 1985, 238, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Schwanzel-Fukuda, M. Origin and migration of luteinizing hormone-releasing hormone neurons in mammals. Microsc. Res. Tech. 1999, 44, 2–10. [Google Scholar] [CrossRef]
- Wirsig-Wiechmann, C.R.; Wiechmann, A.F.; Eisthen, H.L. What defines the nervus terminalis? Neurochemical, developmental, and anatomical criteria. Prog. Brain Res. 2002, 141, 45–58. [Google Scholar]
- Cho, H.-J.; Shan, Y.; Whittington, N.C.; Wray, S. Nasal Placode Development, GnRH Neuronal Migration and Kallmann Syndrome. Front. Cell Dev. Biol. 2019, 7, 121. [Google Scholar] [CrossRef] [Green Version]
- Lehman, M.N.; Merkley, C.M.; Coolen, L.M.; Goodman, R.L. Anatomy of the kisspeptin neural network in mammals. Brain Res. 2010, 1364, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Sonne, J.; Reddy, V.; Lopez-Ojeda, W. Neuroanatomy, Cranial Nerve 0 [Terminal Nerve]; StatPearls: Treasure Island, FL, USA, 24 February 2021. [Google Scholar]
- Skorupskaite, K.; George, J.T.; Anderson, R.A. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod Update 2014, 20, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla, S.L.; Perez, J.G.; Ben-Hamo, M.; Johnson, C.W.; Sanchez, R.E.; Bussi, I.L.; Palmiter, R.D.; Horacio, O. Kisspeptin Neurons in the Arcuate Nucleus of the Hypothalamus Orchestrate Circadian Rhythms and Metabolism. Curr. Biol. 2019, 29, 592–604.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittelman-Smith, M.A.; Williams, H.; Krajewski-Hall, S.J.; McMullen, N.T.; Rance, N.E. Role for kisspeptin/neurokinin B/dynorphin (KNDy) neurons in cutaneous vasodilatation and the estrogen modulation of body temperature. Proc. Natl. Acad. Sci. USA 2012, 109, 19846–19851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rance, N.E.; Dacks, P.A.; Mittelman-Smith, M.A.; Romanovsky, A.A.; Krajewski-Hall, S.J. Modulation of body temperature and LH secretion by hypothalamic KNDy [kisspeptin, neurokinin B and dynorphin] neurons: A novel hypothesis on the mechanism of hot flushes. Front. Neuroendocrinol. 2013, 34, 211–227. [Google Scholar] [CrossRef] [Green Version]
- De Roux, N.; Genin, E.; Carel, J.C.; Matsuda, F.; Chaussain, J.L.; Milgrom, E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. USA 2003, 100, 10972. [Google Scholar] [CrossRef] [Green Version]
- Abbara, A.; Clarke, S.A.; Dhillo, W.S. Clinical Potential of Kisspeptin in Reproductive Health. Trends Mol. Med. 2021, 27, 807–823. [Google Scholar] [CrossRef]
- Liu, X.; Yeo, S.H.; McQuillan, H.J.; Herde, M.K.; Hessler, S.; Cheong, I.; Porteous, R.; Herbison, A.E. Highly redundant neuropeptide volume co-transmission underlying episodic activation of the gnrh neuron dendron. eLife 2021, 10, e62455. [Google Scholar] [CrossRef]
- Mittelman-Smith, M.A.; Williams, H.; Krajewski-Hall, S.J.; Lai, J.; Ciofi, P.; McMullen, N.T.; Rance, N.E. Arcuate Kisspeptin/Neurokinin B/Dynorphin (KNDy) Neurons Mediate the Estrogen Suppression of Gonadotropin Secretion and Body Weight. Endocrinology 2012, 153, 2800–2812. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.T.; Cunningham, M.J.; Rissman, E.F.; Clifton, D.K.; Steiner, R.A. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 2005, 146, 3686–3692. [Google Scholar] [CrossRef]
- Smith, J.T.; Popa, S.M.; Clifton, D.K.; Hoffman, G.E.; Steiner, R.A. Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J. Neurosci. 2006, 26, 6687–6694. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.A.; Singhal, G.; Corrigan, F.; Jaehne, E.J.; Jawahar, M.C.; Breen, J.; Pederson, S.; Baune, B.T. Ceasing exercise induces depression-like, anxiety-like, and impaired cognitive-like behaviours and altered hippocampal gene expression. Brain Res. Bull. 2019, 148, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xie, W.; Li, D.; Shi, L.; Ye, G.; Mao, Y.; Xiong, Y.; Sun, H.; Zheng, F.; Chen, Z. Evaluation of sex-related hormones and semen characteristics in reproductive-aged male COVID-19 patients. J. Med. Virol. 2021, 93, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Çayan, S.; Uğuz, M.; Saylam, B.; Akbay, E. Effect of serum total testosterone and its relationship with other laboratory parameters on the prognosis of coronavirus disease 2019 [COVID-19] in SARS-CoV-2 infected male patients: A cohort study. Aging Male 2020, 23, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, K.; Ravichandran, S.; Krishnan, S.; Radhakrishnan, R.K.; Manickam, N.; Kandasamy, M. Testicular Atrophy and Hypothalamic Pathology in COVID-19: Possibility of the Incidence of Male Infertility and HPG Axis Abnormalities. Reprod. Sci. 2021, 28, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Dutta, S. COVID-19 and hypogonadism: Secondary immune responses rule-over endocrine mechanisms. Hum. Fertil. 2021; ahead-of-print. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Mukhtar, N.; Aljomaiah, A.; Aljamei, H.; Bakhsh, A.; Alsudani, N.; Elsayed, T.; Alrashidi, N.; Fadel, R.; Alqahtani, E.; et al. The Impact of COVID-19 Viral Infection on the Hypothalamic-Pituitary-Adrenal Axis. Endocr. Pract. 2021, 27, 83–89. [Google Scholar] [CrossRef]
- Mayer, C.; Acosta-Martinez, M.; Dubois, S.L.; Wolfe, A.; Radovick, S.; Boehm, U.; Levine, J.E. Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proc. Natl. Acad. Sci. USA 2010, 107, 22693–22698. [Google Scholar] [CrossRef] [Green Version]
- Ding, T.; Zhang, J.; Wang, T.; Cui, P.; Chen, Z.; Jiang, J.; Zhou, S.; Dai, J.; Wang, B.; Yuan, S.; et al. Potential Influence of Menstrual Status and Sex Hormones on Female Severe Acute Respiratory Syndrome Coronavirus 2 Infection: A Cross-sectional Multicenter Study in Wuhan, China. Clin. Infect. Dis. 2021, 72, e240–e248. [Google Scholar] [CrossRef]
- Newson, L.; Lewis, R.; O’Hara, M. Long Covid and menopause—The important role of hormones in Long Covid must be considered. Maturitas 2021, 152, 74. [Google Scholar] [CrossRef]
- WHO. A Clinical Case Definition of Post COVID-19 Condition by a Delphi Consensus, 6 October 2021 [Internet]. 2021. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-Post_COVID-19_condition-Clinical_case_definition-2021.1 (accessed on 1 November 2021).
- Jayasena, C.N.; Comninos, A.N.; Stefanopoulou, E.; Buckley, A.; Narayanaswamy, S.; Izzi-Engbeaya, C.; Abbara, A.; Ratnasabapathy, R.; Mogford, J.; Ng, N.; et al. Neurokinin B administration induces hot flushes in women. Sci. Rep. 2015, 5, 8466. [Google Scholar] [CrossRef] [Green Version]
- Modi, M.; Dhillo, W.S. Neurokinin 3 Receptor Antagonism: A Novel Treatment for Menopausal Hot Flushes. Neuroendocrinology 2019, 109, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.; Riste, L.; Hann, M.; Walther, A.; Mukherjee, A.; Heald, A. Into the looking glass: Post-viral syndrome post COVID-19. Med. Hypotheses 2020, 144, 110055. [Google Scholar] [CrossRef]
- Buoite Stella, A.; Furlanis, G.; Frezza, N.A.; Valentinotti, R.; Ajcevic, M.; Manganotti, P. Autonomic dysfunction in post-COVID patients with and witfhout neurological symptoms: A prospective multidomain observational study. J. Neurol. 2021, 269, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic dysfunction in ‘long COVID’: Rationale, physiology and management strategies. Clin. Med. 2021, 21, e63. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.V.; Latchford, K.J.; Samson, W.K. The paraventricular nucleus of the hypothalamus—A potential target for integrative treatment of autonomic dysfunction. Expert Opin. Ther. Targets 2008, 12, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Dayan, E.; Sklerov, M.; Browner, N. Disrupted hypothalamic functional connectivity in patients with PD and autonomic dysfunction. Neurology 2018, 90, e2051–e2058. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A. A Paradigm for Post-COVID-19 Fatigue Syndrome Analogous to ME/CFS. Front. Neurol. 2021, 12, 701419. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Respiratory sequelae | Dyspnea *, cough, sore throat, altered diffusion capacity, restrictive pattern, obstructive pattern |
| Cardiac sequelae | Palpitations, chest pain, myocarditis |
| Gastrointestinal sequelae | Vomiting/nausea, diarrhea |
| Neurological sequelae | Anosmia, loss of taste, anxiety *, depression, sleeping difficulties, concentration/memory problems *, dizziness, chronic fatigue*, headache |
| Other sequelae | Joint pain, post-exertional malaise *, increased incidence of pain, antihypertensive, and antidepressant drugs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, M.S.; Johansson, A.; Jonsson, J.; Schiöth, H.B. Dissecting the Molecular Mechanisms Surrounding Post-COVID-19 Syndrome and Neurological Features. Int. J. Mol. Sci. 2022, 23, 4275. https://doi.org/10.3390/ijms23084275
Mohamed MS, Johansson A, Jonsson J, Schiöth HB. Dissecting the Molecular Mechanisms Surrounding Post-COVID-19 Syndrome and Neurological Features. International Journal of Molecular Sciences. 2022; 23(8):4275. https://doi.org/10.3390/ijms23084275
Chicago/Turabian StyleMohamed, Mohamed S., Anton Johansson, Jörgen Jonsson, and Helgi B. Schiöth. 2022. "Dissecting the Molecular Mechanisms Surrounding Post-COVID-19 Syndrome and Neurological Features" International Journal of Molecular Sciences 23, no. 8: 4275. https://doi.org/10.3390/ijms23084275
APA StyleMohamed, M. S., Johansson, A., Jonsson, J., & Schiöth, H. B. (2022). Dissecting the Molecular Mechanisms Surrounding Post-COVID-19 Syndrome and Neurological Features. International Journal of Molecular Sciences, 23(8), 4275. https://doi.org/10.3390/ijms23084275