
Identification and Characterization of the MIKC-Type MADS-Box Gene Family in Brassica napus and Its Role in Floral Transition

 , ,
, ,
Abstract
:1. Introduction
2. Results
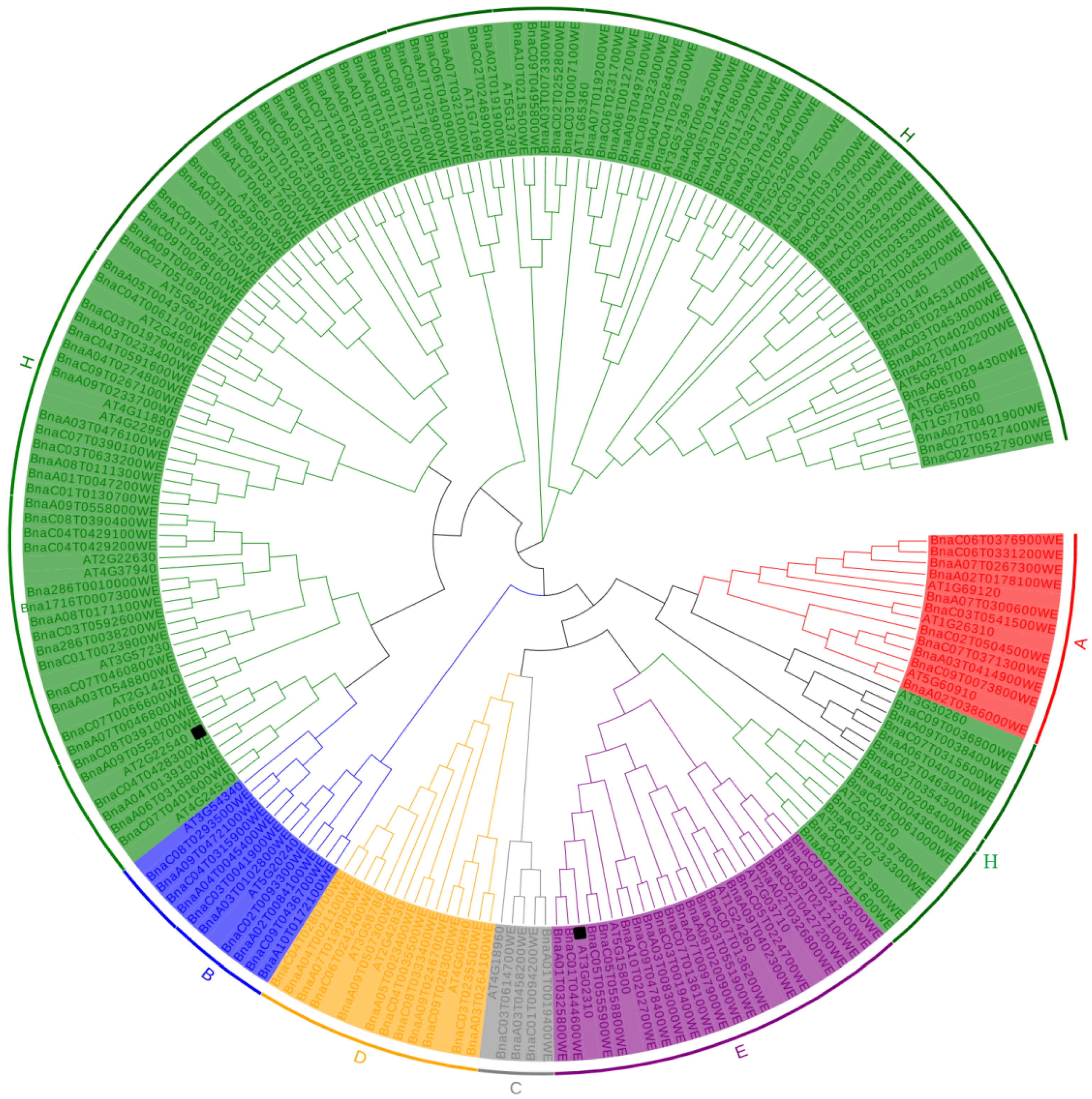
2.1. Identification of Westar MIKC Family Members
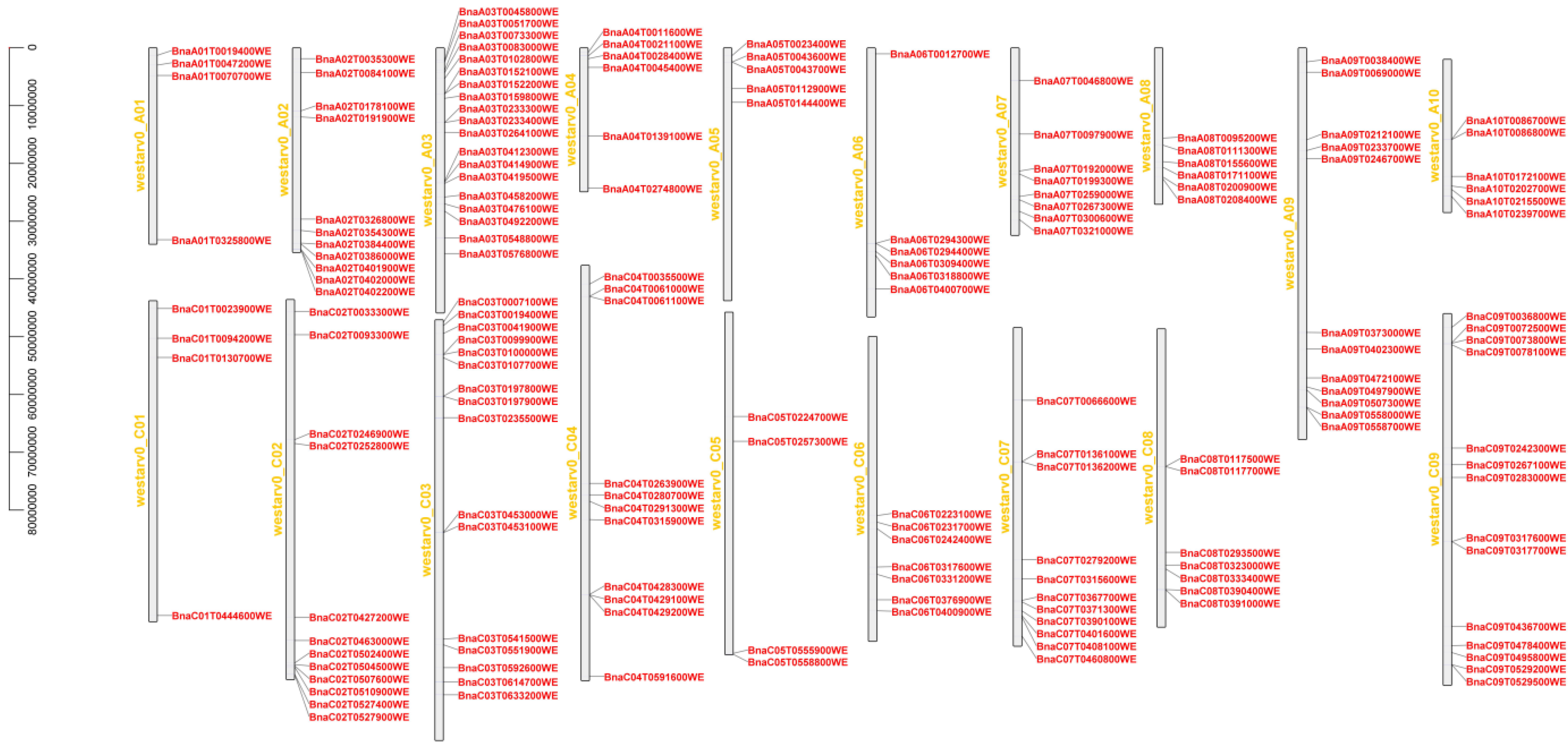
2.2. Chromosome Distributions of the BnaMIKC Genes
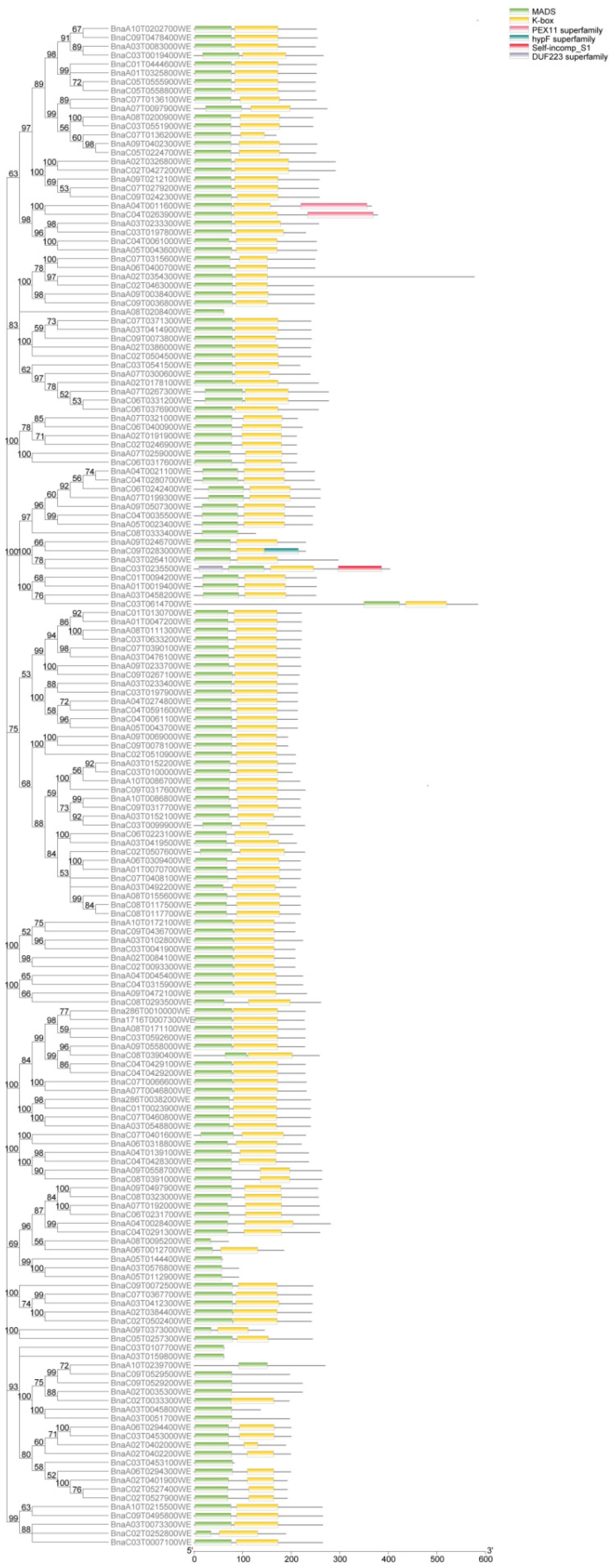
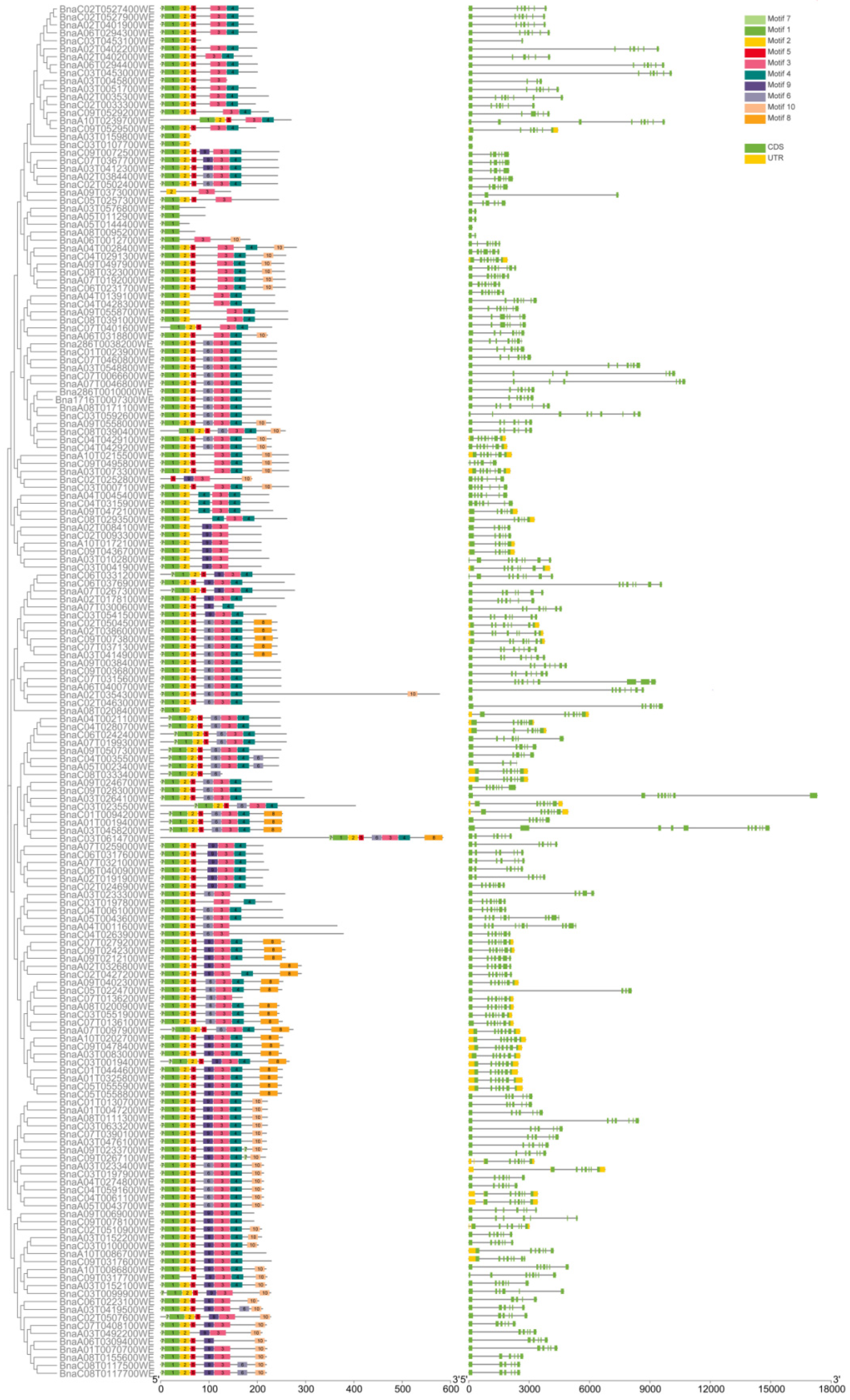
2.3. Protein Domain, Motif, and Gene Structure Analysis
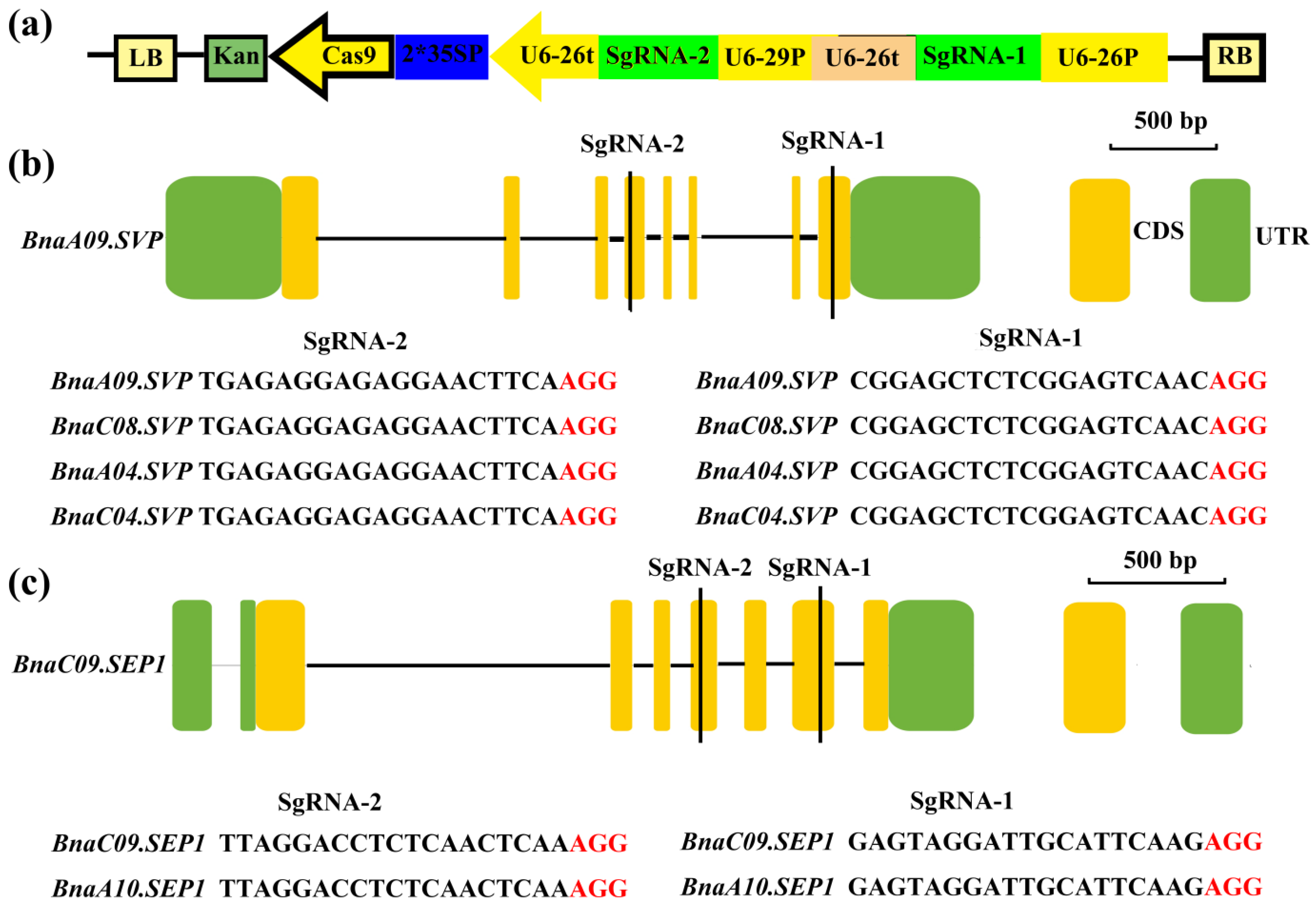
2.4. Design of sgRNAs to Knock Out Homologs of the SVP and SEP1 Genes in B. napus
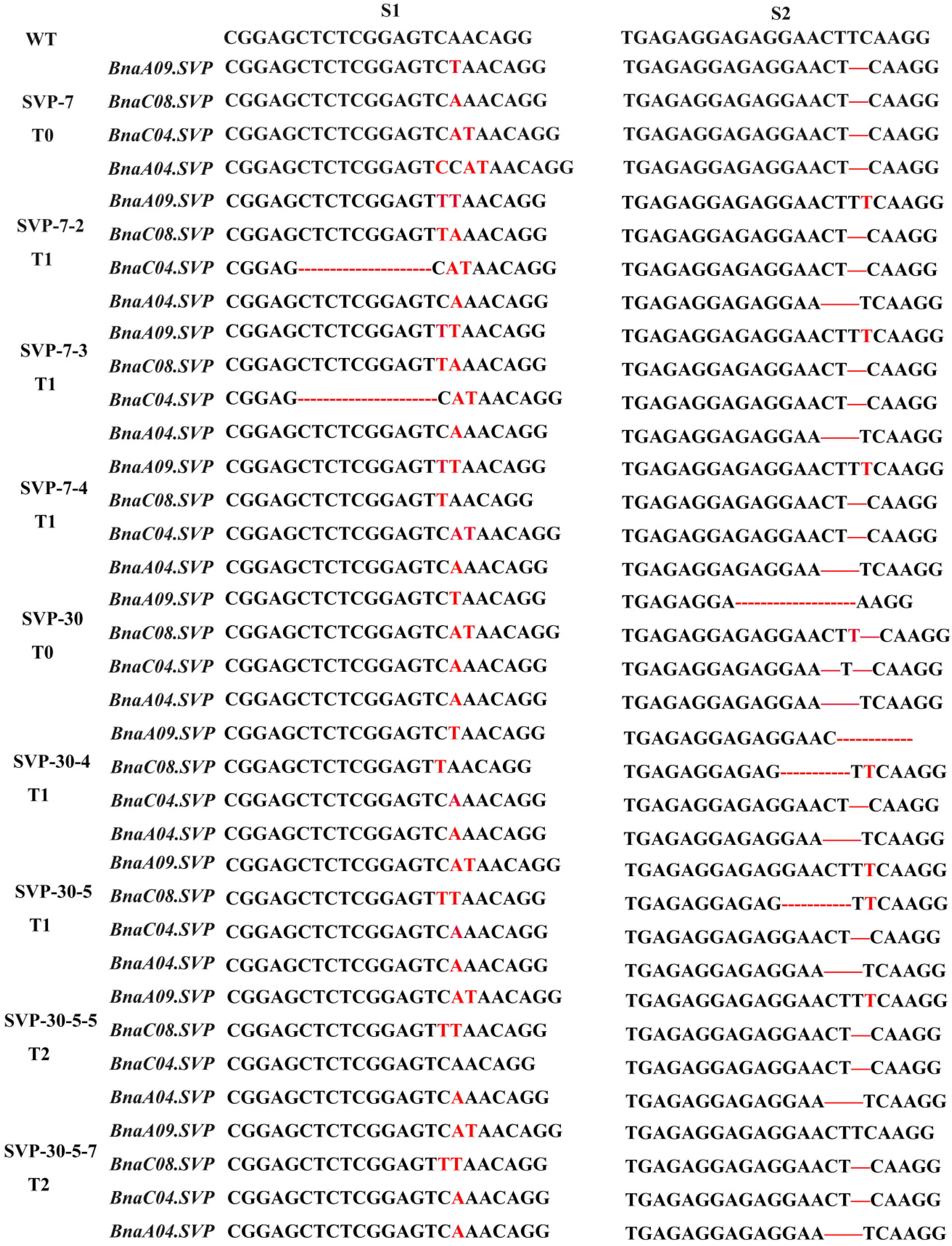
2.5. Identification of BnaSVP and BnaSEP1 Mutations
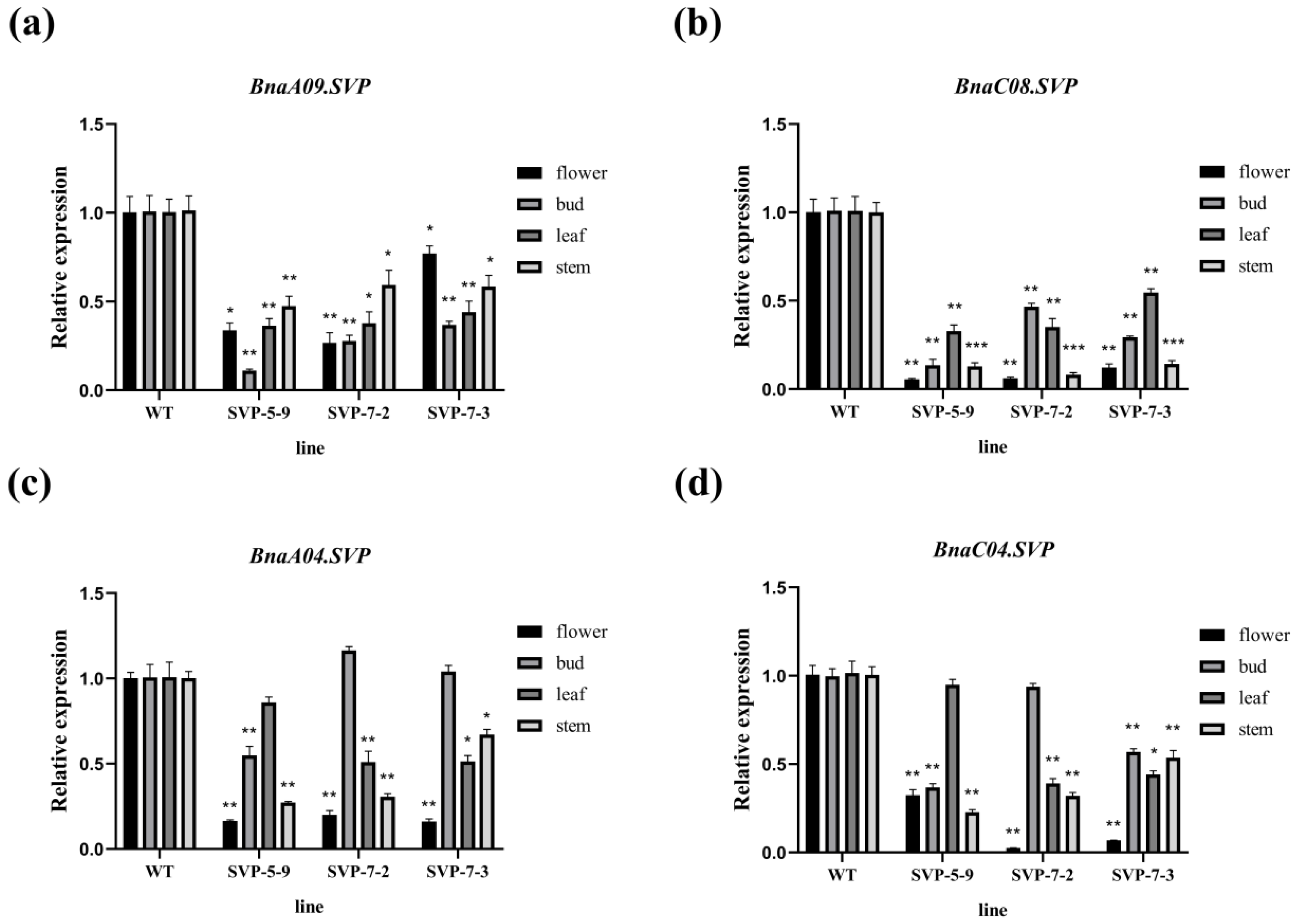
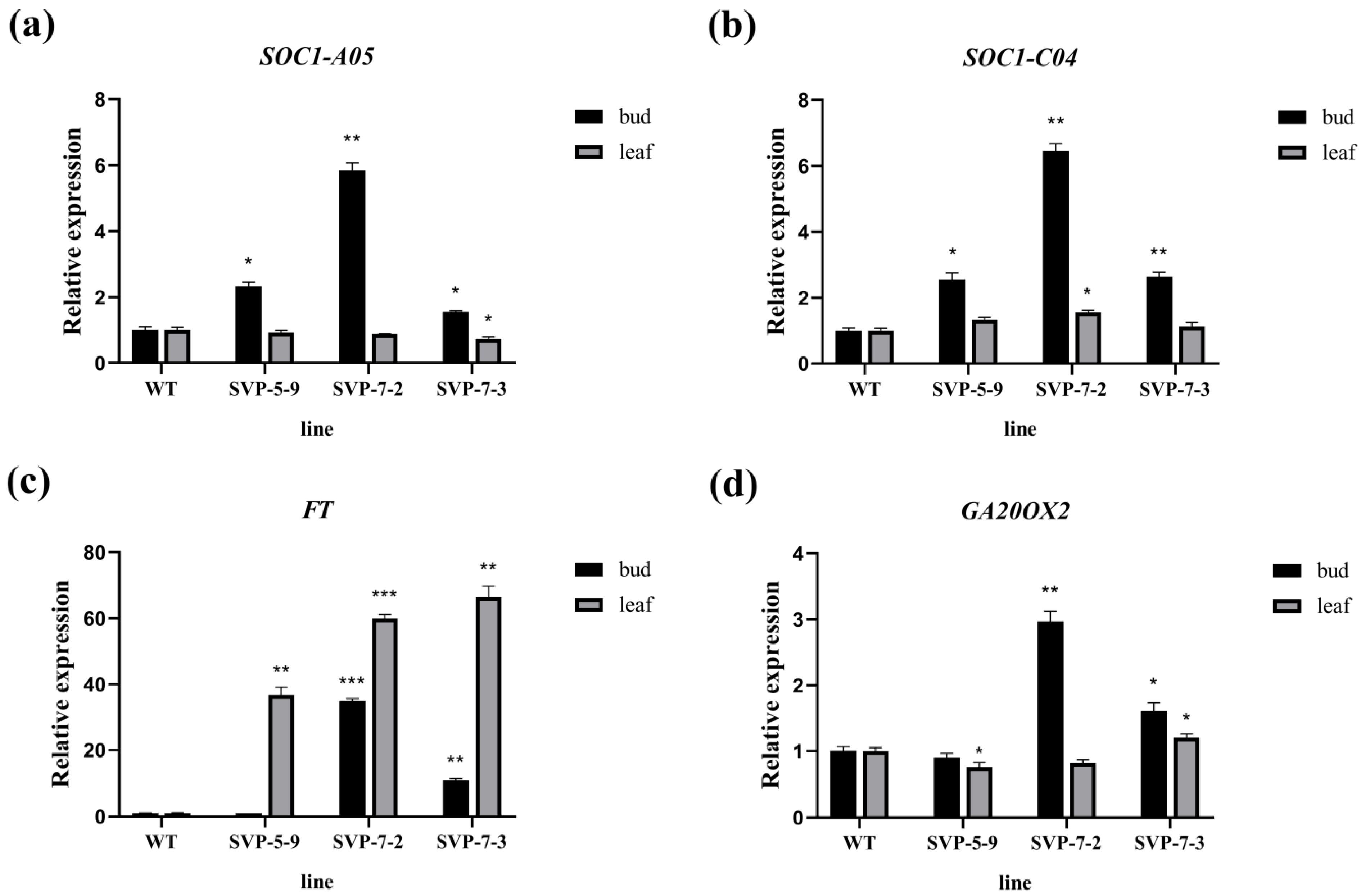
2.6. Expression Analysis of the BnaSVP Gene
3. Discussion
3.1. MIKC Family Genes Provide the Basis for Rapeseed Development
3.2. Homologous Copies of BnaSVP and BnaSEP1 Have Functional Redundancy
3.3. CRISPR/Cas9-Targeted Mutation in BnaSVP Is a Promising Strategy for Early Rapeseed Breeding
3.4. SVP Gene Has Different Functions
4. Materials and Methods
4.1. Plant Materials
4.2. Identification of MICK Family Members in B. napus
4.3. Analysis of MICK Family Gene Characteristics and SVP Protein Interaction Network
4.4. CRISPR/Cas9 Vector Construction and Plant Transformation
4.5. Positive Transgenic-Plant and Mutant Identification
4.6. Phenotypic Observations
4.7. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tasma, I.M.; Lorenzen, L.L.; Green, D.E.; Shoemaker, R.C. Mapping genetic loci for flowering time, maturity, and photoperiod insensitivity in soybean. Mol. Breed. 2001, 8, 25–35. [Google Scholar] [CrossRef]
- Simpson, G.G.; Dean, C. Arabidopsis, the Rosetta stone of flowering time? Science 2002, 296, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouradov, A.; Cremer, F.; Coupland, G. Control of flowering time: Interacting pathways as a basis for diversity. Plant Cell 2002, 14 (Suppl. 1), S111–S130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurie, D.A.; Griffiths, S. Molecular markers for flowering time genes in crop species. In Molecular Techniques in Crop Improvement, 2nd ed.; Mohan Jain, S., Brar, D.S., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 239–263. [Google Scholar] [CrossRef]
- Shindo, C.; Aranzana, M.J.; Lister, C.; Baxter, C.; Nicholls, C.; Nordborg, M.; Dean, C. Role of FRIGIDA and FLOWERING LOCUS C in determining variation in flowering time of Arabidopsis. Plant Physiol. 2005, 138, 1163–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ainsworth, N.E.; Tenaillon, M.I. Superheroes and masterminds of plant domestication. CR Biol. 2016, 339, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.J.; Samaha, R.R.; et al. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Ribas de Pouplana, L.; Martinez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef] [Green Version]
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [Green Version]
- Immink, R.G.; Gadella, T.W., Jr.; Ferrario, S.; Busscher, M.; Angenent, G.C. Analysis of MADS box protein-protein interactions in living plant cells. Proc. Natl. Acad. Sci. USA 2002, 99, 2416–2421. [Google Scholar] [CrossRef] [Green Version]
- Riechmann, J.L.; Wang, M.; Meyerowitz, E.M. DNA-binding properties of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA and AGAMOUS. Nucleic Acids Res. 1996, 24, 3134–3141. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Krizek, B.A.; Meyerowitz, E.M. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. USA 1996, 93, 4793–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Riquelme, J.; Lijavetzky, D.; Martinez-Zapater, J.M.; Carmona, M.J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 2009, 149, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Qu, Y.; Kong, W.; Wang, Q.; Fu, X. Genome-Wide Identification MIKC-Type MADS-Box Gene Family and Their Roles during Development of Floral Buds in Wheel Wingnut (Cyclocarya paliurus). Int. J. Mol. Sci. 2021, 22, 10128. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, Q.; Sun, L.; Du, D.; Cheng, T.; Pan, H.; Yang, W.; Wang, J. Genome-wide identification, characterisation and expression analysis of the MADS-box gene family in Prunus mume. Mol. Genet. Genom. 2014, 289, 903–920. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.E.; Vendramin, E.; Jimenez Tarodo, S.; Verde, I.; Bielenberg, D.G. A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 2015, 15, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; Yu, D.; Wang, D.; Guo, D.; Guo, C. Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol. Biol. Rep. 2013, 40, 3901–3911. [Google Scholar] [CrossRef]
- Parenicova, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, J.; Zhang, J.; Miao, H.; Wang, J.; Gao, P.; Hu, W.; Jia, C.; Wang, Z.; Xu, B.; et al. Genome-wide analysis of banana MADS-box family closely related to fruit development and ripening. Sci. Rep. 2017, 7, 3467. [Google Scholar] [CrossRef]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, W.; Song, X.; Liu, T.; Huang, Z.; Ren, J.; Hou, X.; Li, Y. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genom. 2015, 290, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Yu, D.; Yang, Z.; Li, C.; Qanmber, G.; Li, Y.; Li, J.; Liu, Z.; Lu, L.; Wang, L.; et al. Genome-Wide Identification of the MIKC-Type MADS-Box Gene Family in Gossypium hirsutum L. Unravels Their Roles in Flowering. Front. Plant Sci. 2017, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Lin, H.; Chen, A.; Lau, M.; Jernstedt, J.; Dubcovsky, J. Wheat VRN1, FUL2 and FUL3 play critical and redundant roles in spikelet development and spike determinacy. Development 2019, 146, dev175398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, F.A.J.; Eagles, H.A.; Virgona, J.M.; Martin, P.J.; Condon, J.R.; Angus, J.F. Effect of VRN1 and PPD1 genes on anthesis date and wheat growth. Crop Pasture Sci. 2017, 68, 195–201. [Google Scholar] [CrossRef]
- Yan, L.; Loukoianov, A.; Tranquilli, G.; Helguera, M.; Fahima, T.; Dubcovsky, J. Positional cloning of the wheat vernalization gene VRN1. Proc. Natl. Acad. Sci. USA 2003, 100, 6263–6268. [Google Scholar] [CrossRef] [Green Version]
- Madrid, E.; Chandler, J.W.; Coupland, G. Gene regulatory networks controlled by FLOWERING LOCUS C that confer variation in seasonal flowering and life history. J. Exp. Bot. 2021, 72, 4–14. [Google Scholar] [CrossRef]
- Airoldi, C.A.; McKay, M.; Davies, B. MAF2 Is Regulated by Temperature-Dependent Splicing and Represses Flowering at Low Temperatures in Parallel with FLM. PLoS ONE 2015, 10, e0126516. [Google Scholar] [CrossRef] [Green Version]
- Mateos, J.L.; Madrigal, P.; Tsuda, K.; Rawat, V.; Richter, R.; Romera-Branchat, M.; Fornara, F.; Schneeberger, K.; Krajewski, P.; Coupland, G. Combinatorial activities of SHORT VEGETATIVE PHASE and FLOWERING LOCUS C define distinct modes of flowering regulation in Arabidopsis. Genome Biol. 2015, 16, 31. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Sri, T.; Singh, A.; Mayee, P.; Shivaraj, S.M.; Sharma, P.; Singh, A. SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 influences flowering time, lateral branching, oil quality, and seed yield in Brassica juncea cv. Varuna. Funct. Integr. Genom. 2019, 19, 43–60. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Cheng, Z.J.; Zhang, X.S. Overexpression of TaMADS1, a SEPALLATA-like gene in wheat, causes early flowering and the abnormal development of floral organs in Arabidopsis. Planta 2006, 223, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Balanza, V.; Martinez-Fernandez, I.; Ferrandiz, C. Sequential action of FRUITFULL as a modulator of the activity of the floral regulators SVP and SOC1. J. Exp. Bot. 2014, 65, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaels, S.D.; Ditta, G.; Gustafson-Brown, C.; Pelaz, S.; Yanofsky, M.; Amasino, R.M. AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J. 2003, 33, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Tao, Y.B.; Xu, Z.F. Ectopic expression of Jatropha curcas APETALA1 (JcAP1) caused early flowering in Arabidopsis, but not in Jatropha. PeerJ 2016, 4, e1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Boden, S.A.; Lars, Ø. How can developmental biology help feed a growing population? Development 2019, 146, dev172965. [Google Scholar] [CrossRef] [Green Version]
- Olukolu, B.A.; Tracy, W.F.; Wisser, R.; De Vries, B.; Balint-Kurti, P.J. A Genome-Wide Association Study for Partial Resistance to Maize Common Rust. Phytopathology 2016, 106, 745–751. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, P.; Zhang, X.; Zheng, Q.; Chen, M.; Ge, F.; Li, Z.; Sun, W.; Guan, Z.; Liang, T.; et al. Multi-Locus Genome-Wide Association Study Reveals the Genetic Architecture of Stalk Lodging Resistance-Related Traits in Maize. Front. Plant Sci. 2018, 9, 611. [Google Scholar] [CrossRef]
- Hansen, M.; Kraft, T.; Ganestam, S.; Sall, T.; Nilsson, N.O. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet. Res. 2001, 77, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-genome resequencing of a world-wide collection of rapeseed accessions reveals genetic basis of their ecotype divergence. Mol. Plant 2018, 12, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Raman, H.; Raman, R.; Coombes, N.; Song, J.; Prangnell, R.; Bandaranayake, C.; Tahira, R.; Sundaramoorthi, V.; Killian, A.; Meng, J.; et al. Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola. Plant Cell Environ. 2016, 39, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tu, Y.; Liao, S.; Fu, X.; Lian, X.; He, Y.; Xie, W.; Wang, G. Genome-wide association study of flowering time reveals complex genetic heterogeneity and epistatic interactions in rice. Gene 2021, 770, 145353. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yang, F.; Zhang, J.; Liu, H.; Rahman, S.; Islam, S.; Ma, W.; She, M. Application of CRISPR/Cas9 in Crop Quality Improvement. Int. J. Mol. Sci. 2021, 22, 4206. [Google Scholar] [CrossRef]
- Khalaf, K.; Janowicz, K.; Dyszkiewicz-Konwinska, M.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Jankowski, M.; Machnik, M.; Oleksiewicz, U.; Kocherova, I.; et al. CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes 2020, 11, 921. [Google Scholar] [CrossRef]
- Raffan, S.; Sparks, C.; Huttly, A.; Hyde, L.; Martignago, D.; Mead, A.; Hanley, S.J.; Wilkinson, P.A.; Barker, G.; Edwards, K.J.; et al. Wheat with greatly reduced accumulation of free asparagine in the grain, produced by CRISPR/Cas9 editing of asparagine synthetase gene TaASN2. Plant Biotechnol. J. 2021, 19, 1602–1613. [Google Scholar] [CrossRef]
- Andersson, M.; Turesson, H.; Nicolia, A.; Falt, A.S.; Samuelsson, M.; Hofvander, P. Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Rep. 2017, 36, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Fu, M.; Li, H.; Wang, L.; Liu, R.; Liu, Z.; Zhang, X.; Jin, S. High-oleic acid content, nontransgenic allotetraploid cotton (Gossypium hirsutum L.) generated by knockout of GhFAD2 genes with CRISPR/Cas9 system. Plant Biotechnol. J. 2021, 19, 424–426. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, K.; Li, H.; Han, S.; Meng, Q.; Khan, S.U.; Fan, C.; Xie, K.; Zhou, Y. Precise editing of CLAVATA genes in Brassica napus L. regulates multilocular silique development. Plant Biotechnol. J. 2018, 16, 1322–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaman, Q.U.; Chu, W.; Hao, M.; Shi, Y.; Sun, M.; Sang, S.F.; Mei, D.; Cheng, H.; Liu, J.; Li, C.; et al. CRISPR/Cas9-Mediated Multiplex Genome Editing of JAGGED Gene in Brassica napus L. Biomolecules 2019, 9, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Zhang, L.; Tang, M.; Liu, J.; Liu, H.; Yang, H.; Fan, S.; Terzaghi, W.; Wang, H.; Hua, W. Knockout of two BnaMAX1 homologs by CRISPR/Cas9-targeted mutagenesis improves plant architecture and increases yield in rapeseed (Brassica napus L.). Plant Biotechnol. J. 2020, 18, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Lin, L.; Liu, D.; Wu, D.; Fang, Y.; Wu, J.; Wang, Y. CRISPR/Cas9-Mediated Multiplex Genome Editing of the BnWRKY11 and BnWRKY70 Genes in Brassica napus L. Int. J. Mol. Sci. 2018, 19, 2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramzow, L.; Theissen, G. Phylogenomics of MADS-Box Genes in Plants—Two Opposing Life Styles in One Gene Family. Biology 2013, 2, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Murai, K. Homeotic Genes and the ABCDE Model for Floral Organ Formation in Wheat. Plants 2013, 2, 379–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Hu, K.; Zhang, Z.; Guan, C.; Chen, S.; Hua, W.; Li, J.; Wen, J.; Yi, B.; Shen, J.; et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res. 2016, 23, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Gregis, V.; Sessa, A.; Colombo, L.; Kater, M.M. AGL24, SHORT VEGETATIVE PHASE, and APETALA1 redundantly control AGAMOUS during early stages of flower development in Arabidopsis. Plant Cell 2006, 18, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Wang, C.; Jiao, X.; Zhang, H.; Song, L.; Li, Y.; Gao, C.; Wang, K. Hi-TOM: A platform for high-throughput tracking of mutations induced by CRISPR/Cas systems. Sci. China Life Sci. 2019, 62, 1–7. [Google Scholar] [CrossRef]
- Jang, S.; Torti, S.; Coupland, G. Genetic and spatial interactions between FT, TSF and SVP during the early stages of floral induction in Arabidopsis. Plant J. 2009, 60, 614–625. [Google Scholar] [CrossRef]
- Liu, D.; Yu, L.; Wei, L.; Yu, P.; Wang, J.; Zhao, H.; Zhang, Y.; Zhang, S.; Yang, Z.; Chen, G.; et al. BnTIR: An online transcriptome platform for exploring RNA-seq libraries for oil crop Brassica napus. Plant Biotechnol. J. 2021, 19, 1895–1897. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.H.; Lee, J.S.; Ahn, J.H. A conserved role of SHORT VEGETATIVE PHASE (SVP) in controlling flowering time of Brassica plants. Biochim. Biophys. Acta 2007, 1769, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Andres, F.; Porri, A.; Torti, S.; Mateos, J.; Romera-Branchat, M.; Garcia-Martinez, J.L.; Fornara, F.; Gregis, V.; Kater, M.M.; Coupland, G. SHORT VEGETATIVE PHASE reduces gibberellin biosynthesis at the Arabidopsis shoot apex to regulate the floral transition. Proc. Natl. Acad. Sci. USA 2014, 111, E2760–E2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, S.; Kennedy, A.; Pan, S.; Jermiin, L.S.; Melzer, R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhao, H.; Wang, Y.; Zhang, X.; Zhao, X.; Yuan, Z. Genome-Wide Identification and Expression Analysis of MIKC-Type MADS-Box Gene Family in Punica granatum L. Agronomy 2020, 10, 1197. [Google Scholar] [CrossRef]
- Zhai, Y.; Yu, K.; Cai, S.; Hu, L.; Amoo, O.; Xu, L.; Yang, Y.; Ma, B.; Jiao, Y.; Zhang, C.; et al. Targeted mutagenesis of BnTT8 homologs controls yellow seed coat development for effective oil production in Brassica napus L. Plant Biotechnol. J. 2020, 18, 1153–1168. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Yang, H.; Wu, J.J.; Tang, T.; Liu, K.D.; Dai, C. CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA in Brassica napus. Sci. Rep. 2017, 7, 7489. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Weigel, D.; Beachy, R.N.; Li, J. A proposed regulatory framework for genome-edited crops. Nat. Genet. 2016, 48, 109–111. [Google Scholar] [CrossRef]
- Huang, H.; Cui, T.; Zhang, L.; Yang, Q.; Yang, Y.; Xie, K.; Fan, C.; Zhou, Y. Modifications of fatty acid profile through targeted mutation at BnaFAD2 gene with CRISPR/Cas9-mediated gene editing in Brassica napus. Appl. Genet. 2020, 133, 2401–2411. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.; et al. A Robust CRISPR/Cas9 System for Convenient, High-Efficiency Multiplex Genome Editing in Monocot and Dicot Plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.C.; Kondra, Z.P. A genetic study of growth characters and yield characters of oilseed rape. Euphytica 1978, 27, 177–183. [Google Scholar] [CrossRef]
- Dou, S.; Zhang, T.; Tu, J.; Shen, J.; Yi, B.; Wen, J.; Fu, T.; Dai, C.; Ma, C. Generation of novel self-incompatible Brassica napus by CRISPR/Cas9. Plant Biotechnol. J. 2021, 19, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Gao, Y.; Fan, M.; Yuan, L.; Wu, Z.; Zhang, Q. Overexpression of Chrysanthemum morifolium SVP gene delays blossoming and regulates inflorescence architecture in transgenic Arabidopsis. Can. J. Plant Sci. 2017, 97, 1130–1139. [Google Scholar] [CrossRef]
- Li, Z.M.; Zhang, J.Z.; Mei, L.; Deng, X.X.; Hu, C.G.; Yao, J.L. PtSVP, an SVP homolog from trifoliate orange (Poncirus trifoliata L. Raf.), shows seasonal periodicity of meristem determination and affects flower development in transgenic Arabidopsis and tobacco plants. Plant Mol. Biol. 2010, 74, 129–142. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, H.; Zhang, D.; Yu, D. Ectopic expression of a soybean SVP-like gene in tobacco causes abnormal floral organs and shortens the vegetative phase. Plant Growth Regul. 2016, 80, 345–353. [Google Scholar] [CrossRef]
- Jaudal, M.; Monash, J.; Zhang, L.; Wen, J.; Mysore, K.S.; Macknight, R.; Putterill, J. Overexpression of Medicago SVP genes causes floral defects and delayed flowering in Arabidopsis but only affects floral development in Medicago. J. Exp. Bot. 2014, 65, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.; Kolmos, E.; Folling, M.; Salchert, K.; Storgaard, M.; Jensen, C.S.; Didion, T.; Nielsen, K.K. Two MADS-box genes from perennial ryegrass are regulated by vernalization and involved in the floral transition. Physiol. Plant. 2006, 126, 268–278. [Google Scholar] [CrossRef]
- Xiao, J.; Chen, Y.; Lu, Y.; Liu, Z.; Si, D.; Xu, T.; Sun, L.; Wang, Z.; Yuan, C.; Sun, H.; et al. A natural variation of an SVP MADS-box transcription factor in Triticum petropavlovskyi leads to its ectopic expression and contributes to elongated glume. Mol. Plant 2021, 14, 1408–1411. [Google Scholar] [CrossRef]
- Liu, X.; Sun, Z.; Dong, W.; Wang, Z.; Zhang, L. Expansion and Functional Divergence of the SHORT VEGETATIVE PHASE (SVP) Genes in Eudicots. Genome Biol. Evol. 2018, 10, 3026–3037. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Yoo, S.J.; Park, S.H.; Hwang, I.; Lee, J.S.; Ahn, J.H. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007, 21, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Tate, J.; Mistry, J.; Coggill, P.C.; Sammut, S.J.; Hotz, H.R.; Ceric, G.; Forslund, K.; Eddy, S.R.; Sonnhammer, E.L.; et al. The Pfam protein families database. Nucleic Acids Res. 2008, 36, D281–D288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Xing, H.L.; Dong, L.; Wang, Z.P.; Zhang, H.Y.; Han, C.Y.; Liu, B.; Wang, X.C.; Chen, Q.J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Li, Y.; Li, L.; Du, Z.; Lin, S.; Tian, X.; Li, S.; Yang, B.; Yao, W.; Wang, J. An efficient Agrobacterium-mediated transformation method using hypocotyl as explants for Brassica napus. Mol. Breed. 2020, 40, 1–13. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Target Lines | BnaA09.SVP | BnaC08.SVP | BnaC04.SVP | BnaA04.SVP | Flowering Period Difference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Target 1 | Target 2 | Target 1 | Target 2 | Target 1 | Target 2 | Target 1 | Target 2 | ||
| SVP-W1-2 | I | D+, H | I | D | I | D+ | D | D, H | −20 |
| SVP-W1-4 | I | D+, H | I | D, H | I | I | I, D, H | I, H | −13 |
| SVP-1-5 | D | WT | WT | WT | WT | WT | WT | D, H | 3 |
| SVP-5-4 | I, D+ | D | D | I, D+ | I | I, D | I | D+ | −31 |
| SVP-5-8 | D+ | D | D | D+ | I | D | I | D+ | −31 |
| SVP-5-9 | I | D, H | I | I | I | I | I | D+ | −24 |
| SVP-7-2 | I, G->A | I | I, G->A | D | I, D+ | D | I | D | −29 |
| SVP-7-3 | I, G->A | I, H | I, G->A | D | I, D+ | I | I | D | −23 |
| SVP-7-4 | I, G->A | I, H | G->A | D | I | D | I | D | −17 |
| SVP-8-5 | WT | WT | WT | WT | WT | WT | WT | WT | −2 |
| SVP-11-3 | I | D, H | I | D+ | I | D | I | D | −21 |
| SVP-12-4 | WT | D, I+, H | I | D+ | WT | D, H | WT | D+ | −9 |
| SVP-26-2 | D+, H | D+, H | I, H | WT | I | WT | I, H | WT | −9 |
| SVP-26-9 | D+, H | WT | I, H | WT | I | WT | I, H | WT | −8 |
| SVP-30-5-5 | I | I, H | I, G->A | D | WT | D | I | D | −18 |
| SVP-30-5-7 | I | WT | I, G->A | D, H | I | D | I | D, H | −13 |
| SVP-D-4-10 | I | D | I, H | D | I | I | I, H | I | −8 |
| SVP-D-5-5 | I | D | I | D | I, D+ | I, D+ | I, H | I | −21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, E.; Zhang, Y.; Wang, H.; Jia, Z.; Wang, X.; Wen, J.; Shen, J.; Fu, T.; Yi, B. Identification and Characterization of the MIKC-Type MADS-Box Gene Family in Brassica napus and Its Role in Floral Transition. Int. J. Mol. Sci. 2022, 23, 4289. https://doi.org/10.3390/ijms23084289
Zhou E, Zhang Y, Wang H, Jia Z, Wang X, Wen J, Shen J, Fu T, Yi B. Identification and Characterization of the MIKC-Type MADS-Box Gene Family in Brassica napus and Its Role in Floral Transition. International Journal of Molecular Sciences. 2022; 23(8):4289. https://doi.org/10.3390/ijms23084289
Chicago/Turabian StyleZhou, Enqiang, Yin Zhang, Huadong Wang, Zhibo Jia, Xuejun Wang, Jing Wen, Jinxiong Shen, Tingdong Fu, and Bin Yi. 2022. "Identification and Characterization of the MIKC-Type MADS-Box Gene Family in Brassica napus and Its Role in Floral Transition" International Journal of Molecular Sciences 23, no. 8: 4289. https://doi.org/10.3390/ijms23084289