
Mechanisms Involved in Epileptogenesis in Alzheimer’s Disease and Their Therapeutic Implications



Abstract
:1. Introduction
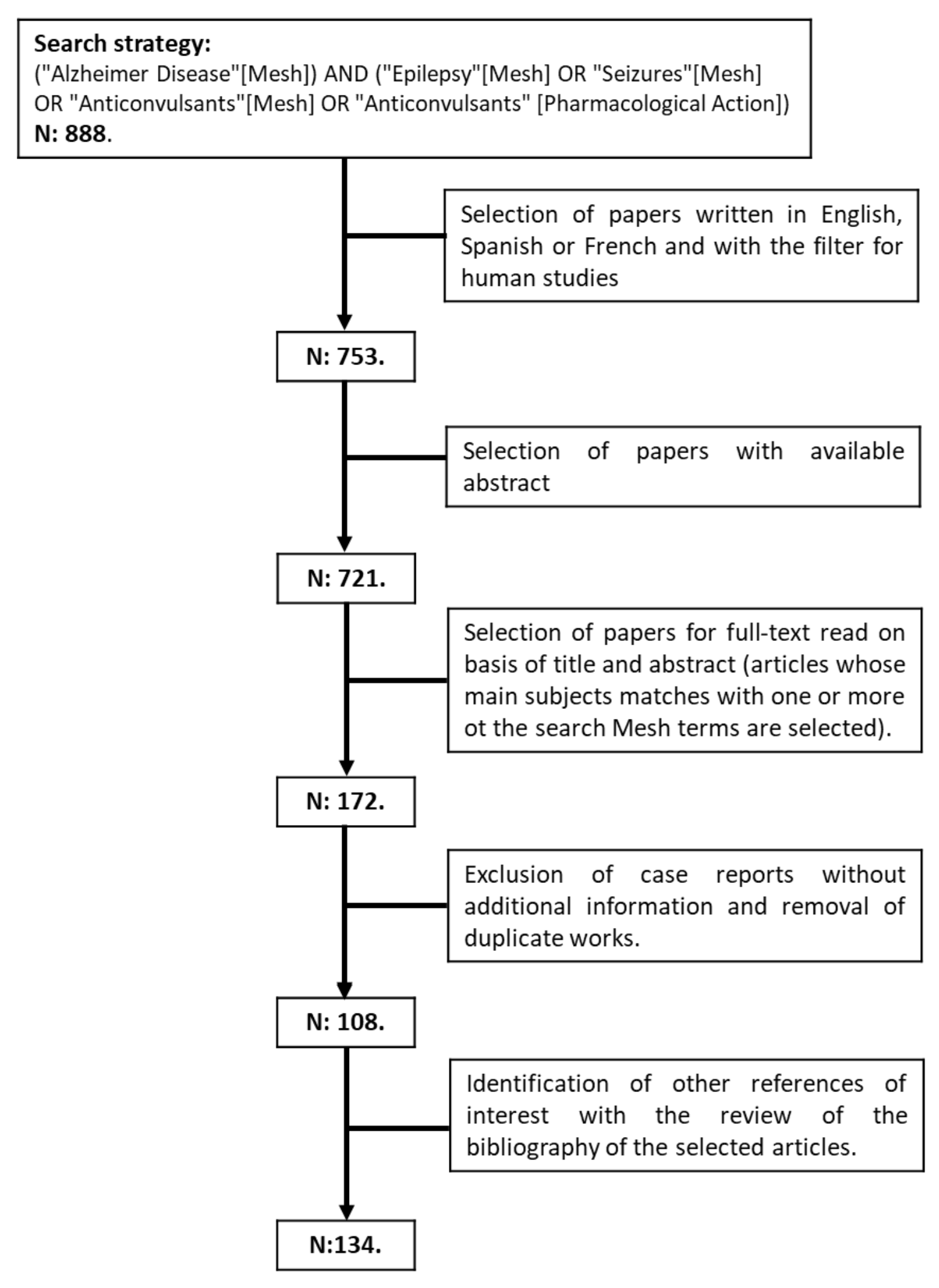
2. Methodology
3. Epilepsy in Alzheimer’s Disease
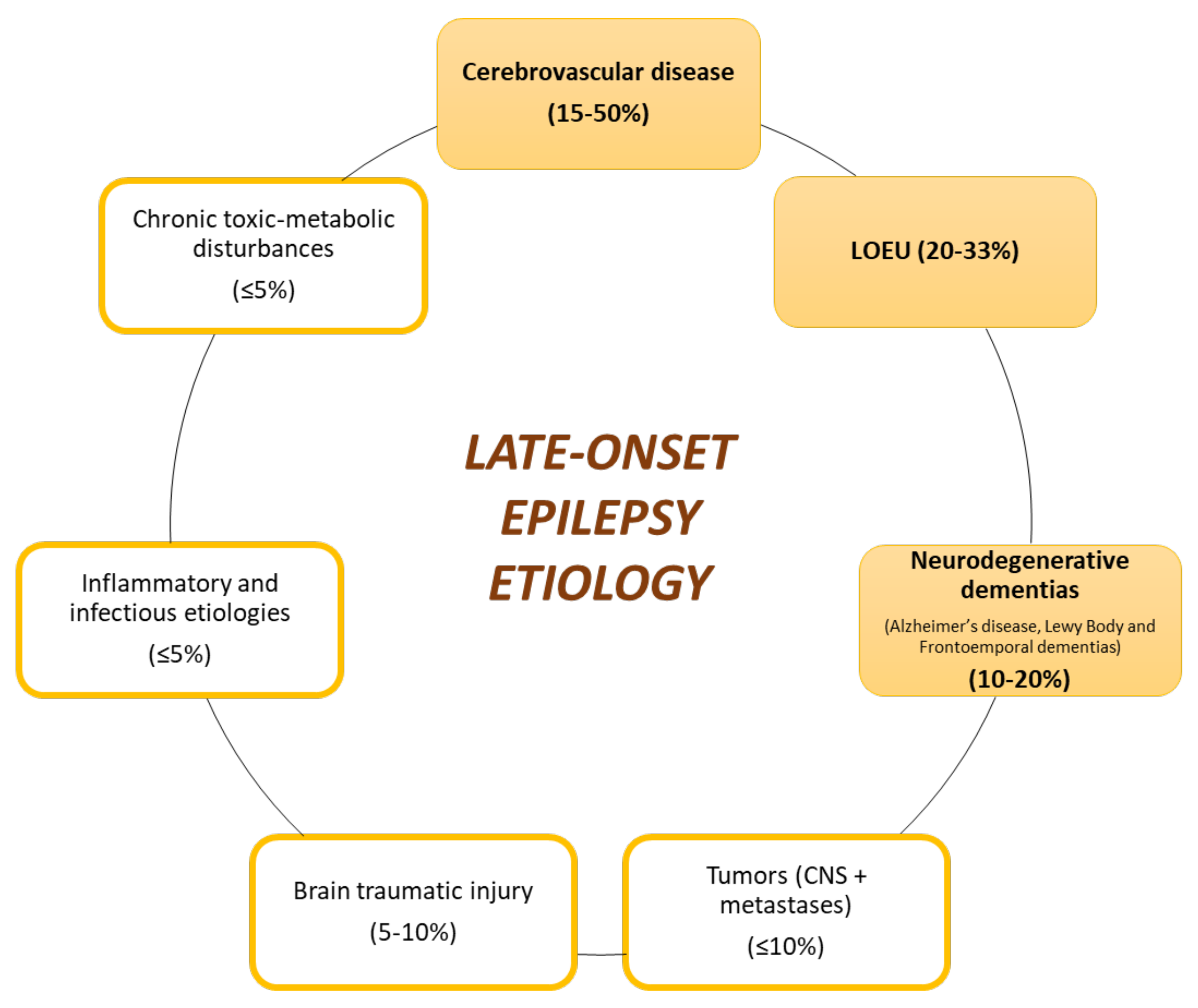
3.1. Late-Onset Epilepsy
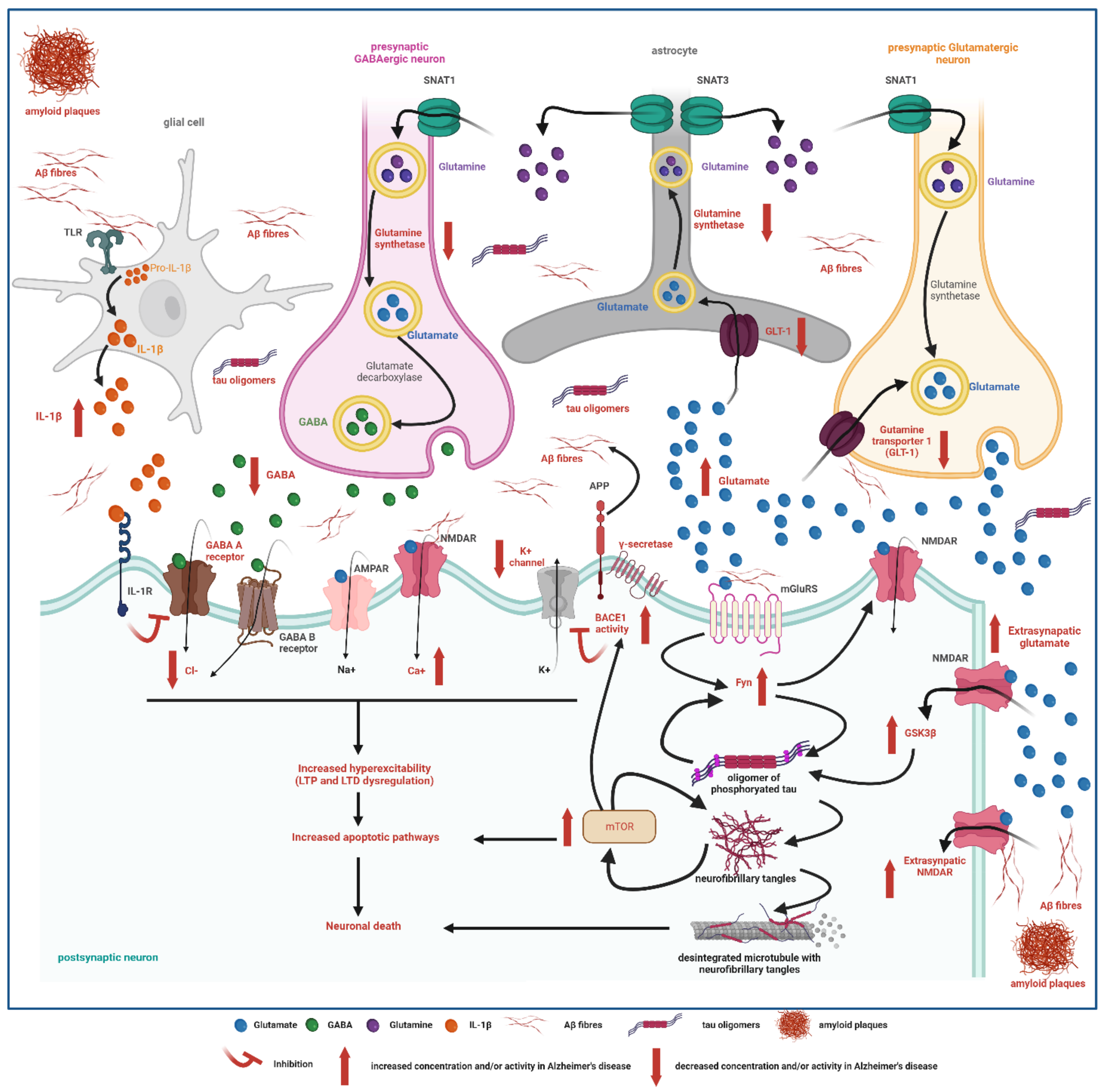
3.2. Epileptogenic Mechanisms in Alzheimer’s Disease
3.2.1. Role of Neurotransmitters in Epileptogenesis
3.2.2. Ion Channel Disruptions
3.2.3. Network Dysfunction
3.3. Amyloid and Tau Promote Hyperexcitability and Facilitate Epileptogenesis
3.3.1. Amyloid (Aβ)
3.3.2. Tau
3.4. Other Mechanisms
3.4.1. Neuroinflammation
3.4.2. mTOR
3.4.3. Apolipoprotein (APOE)
4. Antiseizure Medications (ASMs) in Alzheimer’s Disease
4.1. Treating Epileptic Seizures in AD
4.2. Impact of AD Treatments on Seizure Occurrence and Control
4.3. Antiseizure Medications (ASMs) as Possible Alzheimer’s Disease-Modifying Treatments
4.3.1. SV2A Ligands (Levetiracetam (LEV) and Brivaracetam (BVT))
4.3.2. Sodium Channel Blockers
4.3.3. Calcium Channel Blockers
4.3.4. ASMs with Multiple Mechanisms
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Paradowski, B.; Zagrajek, M.M. Epilepsy in Middle-Aged and Elderly People: A Three-Year Observation. Epileptic Disord. 2005, 7, 91–95. [Google Scholar]
- Stefanidou, M.; Beiser, A.S.; Himali, J.J.; Peng, T.J.; Devinsky, O.; Seshadri, S.; Friedman, D. Bi-Directional Association between Epilepsy and Dementia: The Framingham Heart Study. Neurology 2020, 95, e3241–e3247. [Google Scholar] [CrossRef] [PubMed]
- Vöglein, J.; Ricard, I.; Noachtar, S.; Kukull, W.A.; Dieterich, M.; Levin, J.; Danek, A. Seizures in Alzheimer’s Disease Are Highly Recurrent and Associated with a Poor Disease Course. J. Neurol. 2020, 267, 2941–2948. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, D.; Donniaquio, A.; Mattioli, P.; Massa, F.; Grazzini, M.; Meli, R.; Filippi, L.; Grisanti, S.; Famà, F.; Terzaghi, M.; et al. Epilepsy in Neurodegenerative Dementias: A Clinical, Epidemiological, and EEG Study. J. Alzheimer’s Dis. 2020, 74, 865–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asadollahi, M.; Atazadeh, M.; Noroozian, M. Seizure in Alzheimer’s Disease: An Underestimated Phenomenon. Am. J. Alzheimer’s Dis. Other Dement. 2019, 34, 81–88. [Google Scholar] [CrossRef]
- Beagle, A.J.; Darwish, S.M.; Ranasinghe, K.G.; La, A.L.; Karageorgiou, E.; Vossel, K.A. Relative Incidence of Seizures and Myoclonus in Alzheimer’s Disease, Dementia with Lewy Bodies, and Frontotemporal Dementia. J. Alzheimer’s Dis. 2017, 60, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Süße, M.; Hamann, L.; Flöel, A.; von Podewils, F. Nonlesional Late-Onset Epilepsy: Semiology, EEG, Cerebrospinal Fluid, and Seizure Outcome Characteristics. Epilepsy Behav. 2019, 91, 75–80. [Google Scholar] [CrossRef]
- Born, H.A. Seizures in Alzheimer’s Disease. Neuroscience 2015, 286, 251–263. [Google Scholar] [CrossRef]
- Cook, M.; Baker, N.; Lanes, S.; Bullock, R.; Wentworth, C.; Arrighi, H.M. Incidence of Stroke and Seizure in Alzheimer’s Disease Dementia. Age Ageing 2015, 44, 695–699. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, F.S.; Baldacci, F.; Dini, E.; Tognoni, G.; Bonuccelli, U. Epilepsy Occurrence in Patients with Alzheimer’s Disease: Clinical Experience in a Tertiary Dementia Center. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2016, 37, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Subota, A.; Pham, T.; Jetté, N.; Sauro, K.; Lorenzetti, D.; Holroyd-Leduc, J. The Association between Dementia and Epilepsy: A Systematic Review and Meta-Analysis. Epilepsia 2017, 58, 962–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romoli, M.; Sen, A.; Parnetti, L.; Calabresi, P.; Costa, C. Amyloid-β: A Potential Link between Epilepsy and Cognitive Decline. Nat. Rev. Neurol. 2021, 17, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Horváth, A.; Szűcs, A.; Hidasi, Z.; Csukly, G.; Barcs, G.; Kamondi, A. Prevalence, Semiology, and Risk Factors of Epilepsy in Alzheimer’s Disease: An Ambulatory EEG Study. J. Alzheimer’s Dis. 2018, 63, 1045–1054. [Google Scholar] [CrossRef] [Green Version]
- Paudel, Y.N.; Angelopoulou, E.; Jones, N.C.; O’Brien, T.J.; Kwan, P.; Piperi, C.; Othman, I.; Shaikh, M.F. Tau Related Pathways as a Connecting Link between Epilepsy and Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 4199–4212. [Google Scholar] [CrossRef]
- Noebels, J. A Perfect Storm: Converging Paths of Epilepsy and Alzheimer’s Dementia Intersect in the Hippocampal Formation. Epilepsia 2011, 52 (Suppl. 1), 39–46. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.L.; Krauss, G.L.; Kucharska-Newton, A.; Albert, M.S.; Brandt, J.; Walker, K.A.; Yasar, S.; Knopman, D.S.; Vossel, K.A.; Gottesman, R.F. Dementia in Late-Onset Epilepsy: The Atherosclerosis Risk in Communities Study. Neurology 2020, 95, e3248–e3256. [Google Scholar] [CrossRef]
- Keret, O.; Hoang, T.D.; Xia, F.; Rosen, H.J.; Yaffe, K. Association of Late-Onset Unprovoked Seizures of Unknown Etiology With the Risk of Developing Dementia in Older Veterans. JAMA Neurol. 2020, 77, 710–715. [Google Scholar] [CrossRef]
- Carter, M.D.; Weaver, D.F.; Joudrey, H.R.; Carter, A.O.; Rockwood, K. Epilepsy and Antiepileptic Drug Use in Elderly People as Risk Factors for Dementia. J. Neurol. Sci. 2007, 252, 169–172. [Google Scholar] [CrossRef]
- Ophir, K.; Ran, B.; Felix, B.; Amir, G. Ten Year Cumulative Incidence of Dementia after Late Onset Epilepsy of Unknown Etiology. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2021, 86, 247–251. [Google Scholar] [CrossRef]
- Johnson, E.L.; Krauss, G.L.; Lee, A.K.; Schneider, A.L.C.; Dearborn, J.L.; Kucharska-Newton, A.M.; Huang, J.; Alonso, A.; Gottesman, R.F. Association Between Midlife Risk Factors and Late-Onset Epilepsy: Results From the Atherosclerosis Risk in Communities Study. JAMA Neurol. 2018, 75, 1375–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.; Romoli, M.; Liguori, C.; Farotti, L.; Eusebi, P.; Bedetti, C.; Siliquini, S.; Cesarini, E.N.; Romigi, A.; Mercuri, N.B.; et al. Alzheimer’s Disease and Late-Onset Epilepsy of Unknown Origin: Two Faces of Beta Amyloid Pathology. Neurobiol. Aging 2019, 73, 61–67. [Google Scholar] [CrossRef]
- Toniolo, S.; Sen, A.; Husain, M. Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9318. [Google Scholar] [CrossRef]
- Kazim, S.F.; Seo, J.H.; Bianchi, R.; Larson, C.S.; Sharma, A.; Wong, R.K.S.; Gorbachev, K.Y.; Pereira, A.C. Neuronal Network Excitability in Alzheimer’s Disease: The Puzzle of Similar versus Divergent Roles of Amyloid β and Tau. eNeuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Gourmaud, S.; Shou, H.; Irwin, D.J.; Sansalone, K.; Jacobs, L.M.; Lucas, T.H.; Marsh, E.D.; Davis, K.A.; Jensen, F.E.; Talos, D.M. Alzheimer-like Amyloid and Tau Alterations Associated with Cognitive Deficit in Temporal Lobe Epilepsy. Brain 2020, 143, 191–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, D.; Honig, L.S.; Scarmeas, N. Seizures and Epilepsy in Alzheimer’s Disease. CNS Neurosci. Ther. 2012, 18, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, R.; Liu, J.Y.W.; Sisodiya, S.M.; Thom, M. A Comparative Study of the Dentate Gyrus in Hippocampal Sclerosis in Epilepsy and Dementia. Neuropathol. Appl. Neurobiol. 2014, 40, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larner, A.J. Epileptic Seizures in AD Patients. Neuromolecular Med. 2010, 12, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Horváth, A.; Szűcs, A.; Barcs, G.; Noebels, J.L.; Kamondi, A. Epileptic Seizures in Alzheimer Disease: A Review. Alzheimer Dis. Assoc. Disord. 2016, 30, 186–192. [Google Scholar] [CrossRef]
- Altuna, M.; Giménez, S.; Fortea, J. Epilepsy in Down Syndrome: A Highly Prevalent Comorbidity. J. Clin. Med. 2021, 10, 2776. [Google Scholar] [CrossRef]
- Garg, N.; Joshi, R.; Medhi, B. Cracking Novel Shared Targets between Epilepsy and Alzheimer’s Disease: Need of the Hour. Rev. Neurosci. 2018, 29, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lavrencic, L.; Radford, K.; Booth, A.; Yoshimura, S.; Anstey, K.J.; Anderson, C.S.; Peters, R. Systematic Review of Coexistent Epileptic Seizures and Alzheimer’s Disease: Incidence and Prevalence. J. Am. Geriatr. Soc. 2021, 69, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Aller-Alvarez, J.S.; Menendez-Gonzalez, M.; Ribacoba-Montero, R.; Salvado, M.; Vega, V.; Suarez-Moro, R.; Sueiras, M.; Toledo, M.; Salas-Puig, J.; Alvarez-Sabin, J. Myoclonic Epilepsy in Down Syndrome and Alzheimer Disease. Neurologia 2017, 32, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Tait, L.; Lopes, M.A.; Stothart, G.; Baker, J.; Kazanina, N.; Zhang, J.; Goodfellow, M. A Large-Scale Brain Network Mechanism for Increased Seizure Propensity in Alzheimer’s Disease. PLoS Comput. Biol. 2021, 17, e1009252. [Google Scholar] [CrossRef]
- Li, B.-Y.; Chen, S.-D. Potential Similarities in Temporal Lobe Epilepsy and Alzheimer’s Disease: From Clinic to Pathology. Am. J. Alzheimer’s Dis. Other Dement. 2015, 30, 723–728. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Saccaro, L.F.; Busceti, C.L.; Biagioni, F.; Fornai, F. Epilepsy and Alzheimer’s Disease: Potential Mechanisms for an Association. Brain Res. Bull. 2020, 160, 107–120. [Google Scholar] [CrossRef]
- Hommet, C.; Mondon, K.; Camus, V.; De Toffol, B.; Constans, T. Epilepsy and Dementia in the Elderly. Dement. Geriatr. Cogn. Disord. 2008, 25, 293–300. [Google Scholar] [CrossRef]
- Zarea, A.; Charbonnier, C.; Rovelet-Lecrux, A.; Nicolas, G.; Rousseau, S.; Borden, A.; Pariente, J.; Le Ber, I.; Pasquier, F.; Formaglio, M.; et al. Seizures in Dominantly Inherited Alzheimer Disease. Neurology 2016, 87, 912–919. [Google Scholar] [CrossRef]
- Harris, S.S.; Wolf, F.; De Strooper, B.; Busche, M.A. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer’s Disease. Neuron 2020, 107, 417–435. [Google Scholar] [CrossRef]
- Larner, A.J. Presenilin-1 Mutation Alzheimer’s Disease: A Genetic Epilepsy Syndrome? Epilepsy Behav. 2011, 21, 20–22. [Google Scholar] [CrossRef]
- Lozsadi, D.A.; Larner, A.J. Prevalence and Causes of Seizures at the Time of Diagnosis of Probable Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 121–124. [Google Scholar] [CrossRef]
- Picco, A.; Archetti, S.; Ferrara, M.; Arnaldi, D.; Piccini, A.; Serrati, C.; di Lorenzo, D.; Morbelli, S.; Nobili, F. Seizures Can Precede Cognitive Symptoms in Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 737–742. [Google Scholar] [CrossRef]
- Vöglein, J.; Noachtar, S.; McDade, E.; Quaid, K.A.; Salloway, S.; Ghetti, B.; Noble, J.; Berman, S.; Chhatwal, J.; Mori, H.; et al. Seizures as an Early Symptom of Autosomal Dominant Alzheimer’s Disease. Neurobiol. Aging 2019, 76, 18–23. [Google Scholar] [CrossRef]
- Cretin, B.; Blanc, F.; Gaultier, C.; Sellal, F. Epileptic Amnesic Syndrome Revealing Alzheimer’s Disease. Epilepsy Res. 2012, 102, 206–209. [Google Scholar] [CrossRef]
- Kang, J.-Q. Epileptic Mechanisms Shared by Alzheimer’s Disease: Viewed via the Unique Lens of Genetic Epilepsy. Int. J. Mol. Sci. 2021, 22, 7133. [Google Scholar] [CrossRef]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and Predictors of Seizures in Patients with Alzheimer’s Disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef]
- Bernardi, S.; Scaldaferri, N.; Vanacore, N.; Trebbastoni, A.; Francia, A.; D’Amico, A.; Prencipe, M. Seizures in Alzheimer’s Disease: A Retrospective Study of a Cohort of Outpatients. Epileptic Disord. 2010, 12, 16–21. [Google Scholar] [CrossRef]
- Cheng, C.-H.; Liu, C.-J.; Ou, S.-M.; Yeh, C.-M.; Chen, T.-J.; Lin, Y.-Y.; Wang, S.-J. Incidence and Risk of Seizures in Alzheimer’s Disease: A Nationwide Population-Based Cohort Study. Epilepsy Res. 2015, 115, 63–66. [Google Scholar] [CrossRef]
- Zelano, J.; Brigo, F.; Garcia-Patek, S. Increased Risk of Epilepsy in Patients Registered in the Swedish Dementia Registry. Eur. J. Neurol. 2020, 27, 129–135. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Jin, S.; He, F.; Emond, J.A.; Raman, R.; Thomas, R.G.; Sano, M.; Quinn, J.F.; Tariot, P.N.; Galasko, D.R.; et al. Incidence of New-Onset Seizures in Mild to Moderate Alzheimer Disease. Arch. Neurol. 2012, 69, 368–372. [Google Scholar] [CrossRef]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic Activity in Alzheimer’s Disease: Causes and Clinical Relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, A.; Abel, E.; Fraser, M.R.; Ryan, N.S.; Jiménez, D.A.; Koriath, C.; Chávez-Gutiérrez, L.; Ansorge, O.; Mummery, C.J.; Lashley, T.; et al. A Novel Presenilin 1 Duplication Mutation (Ile168dup) Causing Alzheimer’s Disease Associated with Myoclonus, Seizures and Pyramidal Features. Neurobiol. Aging 2021, 103, 137.e1–137.e5. [Google Scholar] [CrossRef] [PubMed]
- Cortini, F.; Cantoni, C.; Villa, C. Epileptic Seizures in Autosomal Dominant Forms of Alzheimer’s Disease. Seizure 2018, 61, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fray, S.; Rassas, A.; Messaoud, T.; Belal, S. Refractory Epilepsy in PSEN 1 Mutation (I83T). Neurocase 2020, 26, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Cretin, B.; Sellal, F.; Philippi, N.; Bousiges, O.; Di Bitonto, L.; Martin-Hunyadi, C.; Blanc, F. Epileptic Prodromal Alzheimer’s Disease, a Retrospective Study of 13 New Cases: Expanding the Spectrum of Alzheimer’s Disease to an Epileptic Variant? J. Alzheimer’s Dis. 2016, 52, 1125–1133. [Google Scholar] [CrossRef]
- Liedorp, M.; Stam, C.J.; van der Flier, W.M.; Pijnenburg, Y.A.L.; Scheltens, P. Prevalence and Clinical Significance of Epileptiform EEG Discharges in a Large Memory Clinic Cohort. Dement. Geriatr. Cogn. Disord. 2010, 29, 432–437. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Chen, T.-S.; Huang, C.-W. Dementia with Non-Convulsive Seizures: A Case Report. J. Int. Med. Res. 2021, 49, 3000605211062453. [Google Scholar] [CrossRef]
- Cretin, B.; Philippi, N.; Bousiges, O.; Dibitonto, L.; Sellal, F.; Martin-Hunyadi, C.; Blanc, F. Do We Know How to Diagnose Epilepsy Early in Alzheimer’s Disease? Rev. Neurol. 2017, 173, 374–380. [Google Scholar] [CrossRef]
- Tang, M.; Ryman, D.C.; McDade, E.; Jasielec, M.S.; Buckles, V.D.; Cairns, N.J.; Fagan, A.M.; Goate, A.; Marcus, D.S.; Xiong, C.; et al. Neurological Manifestations of Autosomal Dominant Familial Alzheimer’s Disease: A Comparison of the Published Literature with the Dominantly Inherited Alzheimer Network Observational Study (DIAN-OBS). Lancet Neurol. 2016, 15, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Nimmrich, V.; Draguhn, A.; Axmacher, N. Neuronal Network Oscillations in Neurodegenerative Diseases. Neuromol. Med. 2015, 17, 270–284. [Google Scholar] [CrossRef]
- Horváth, A.; Szűcs, A.; Barcs, G.; Kamondi, A. Sleep EEG Detects Epileptiform Activity in Alzheimer’s Disease with High Sensitivity. J. Alzheimer’s Dis. 2017, 56, 1175–1183. [Google Scholar] [CrossRef]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent Hippocampal Seizures and Spikes Identified by Foramen Ovale Electrodes in Alzheimer’s Disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and Impact of Subclinical Epileptiform Activity in Alzheimer’s Disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef]
- Liguori, C.; Spanetta, M.; Romoli, M.; Placidi, F.; Nardi Cesarini, E.; Mercuri, N.B.; Costa, C. Sleep Disorders and Late-Onset Epilepsy of Unknown Origin: Understanding New Trajectories to Brain Amyloidopathy. Mech. Ageing Dev. 2021, 194, 111434. [Google Scholar] [CrossRef]
- Lam, A.D.; Sarkis, R.A.; Pellerin, K.R.; Jing, J.; Dworetzky, B.A.; Hoch, D.B.; Jacobs, C.S.; Lee, J.W.; Weisholtz, D.S.; Zepeda, R.; et al. Association of Epileptiform Abnormalities and Seizures in Alzheimer Disease. Neurology 2020, 95, e2259–e2270. [Google Scholar] [CrossRef]
- Milikovsky, D.Z.; Ofer, J.; Senatorov, V.V.J.; Friedman, A.R.; Prager, O.; Sheintuch, L.; Elazari, N.; Veksler, R.; Zelig, D.; Weissberg, I.; et al. Paroxysmal Slow Cortical Activity in Alzheimer’s Disease and Epilepsy Is Associated with Blood-Brain Barrier Dysfunction. Sci. Transl. Med. 2019, 11, eaaw8954. [Google Scholar] [CrossRef]
- Brunetti, V.; D’Atri, A.; Della Marca, G.; Vollono, C.; Marra, C.; Vita, M.G.; Scarpelli, S.; De Gennaro, L.; Rossini, P.M. Subclinical Epileptiform Activity during Sleep in Alzheimer’s Disease and Mild Cognitive Impairment. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2020, 131, 1011–1018. [Google Scholar] [CrossRef]
- Sen, A.; Jette, N.; Husain, M.; Sander, J.W. Epilepsy in Older People. Lancet 2020, 395, 735–748. [Google Scholar] [CrossRef]
- Powell, G.; Ziso, B.; Larner, A.J. The Overlap between Epilepsy and Alzheimer’s Disease and the Consequences for Treatment. Expert Rev. Neurother. 2019, 19, 653–661. [Google Scholar] [CrossRef]
- Baker, J.; Libretto, T.; Henley, W.; Zeman, A. A Longitudinal Study of Epileptic Seizures in Alzheimer’s Disease. Front. Neurol. 2019, 10, 1266. [Google Scholar] [CrossRef] [Green Version]
- DiFrancesco, J.C.; Tremolizzo, L.; Polonia, V.; Giussani, G.; Bianchi, E.; Franchi, C.; Nobili, A.; Appollonio, I.; Beghi, E.; Ferrarese, C. Adult-Onset Epilepsy in Presymptomatic Alzheimer’s Disease: A Retrospective Study. J. Alzheimer’s Dis. 2017, 60, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Kiss, M.; Szucs, A.; Kamondi, A. Precuneus-Dominant Degeneration of Parietal Lobe Is at Risk of Epilepsy in Mild Alzheimer’s Disease. Front. Neurol. 2019, 10, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.; Robertson, N.P. Seizures in Alzheimer’s Disease: Is There More beneath the Surface? J. Neurol. 2018, 265, 226–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, A.A.; Papp, A.; Zsuffa, J.; Szucs, A.; Luckl, J.; Radai, F.; Nagy, F.; Hidasi, Z.; Csukly, G.; Barcs, G.; et al. Subclinical Epileptiform Activity Accelerates the Progression of Alzheimer’s Disease: A Long-Term EEG Study. Clin. Neurophysiol. 2021, 132, 1982–1989. [Google Scholar] [CrossRef]
- Nardi Cesarini, E.; Babiloni, C.; Salvadori, N.; Farotti, L.; Del Percio, C.; Pascarelli, M.T.; Noce, G.; Lizio, R.; Da Re, F.; Isella, V.; et al. Late-Onset Epilepsy With Unknown Etiology: A Pilot Study on Neuropsychological Profile, Cerebrospinal Fluid Biomarkers, and Quantitative EEG Characteristics. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Rohracher, A.; Kalss, G.; Kuchukhidze, G.; Neuray, C.; Leitinger, M.; Höfler, J.; Kreidenhuber, R.; Rossini, F.; Volna, K.; Mauritz, M.; et al. New Anti-Seizure Medication for Elderly Epilepsy Patients—A Critical Narrative Review. Expert Opin. Pharmacother. 2021, 22, 621–634. [Google Scholar] [CrossRef]
- Wojewodka, G.; McKinlay, A.; Ridsdale, L. Best Care for Older People with Epilepsy: A Scoping Review. Seizure 2021, 85, 70–89. [Google Scholar] [CrossRef]
- Subota, A.; Jetté, N.; Josephson, C.B.; McMillan, J.; Keezer, M.R.; Gonzalez-Izquierdo, A.; Holroyd-Leduc, J. Risk Factors for Dementia Development, Frailty, and Mortality in Older Adults with Epilepsy—A Population-Based Analysis. Epilepsy Behav. 2021, 120, 108006. [Google Scholar] [CrossRef]
- Feyissa, A.M.; Hasan, T.F.; Meschia, J.F. Stroke-Related Epilepsy. Eur. J. Neurol. 2019, 26, 18-e3. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Thacker, E.L.; Longstreth, W.T.J.; Elkind, M.S.V.; Boehme, A.K. Cognitive Decline in Older Adults with Epilepsy: The Cardiovascular Health Study. Epilepsia 2021, 62, 85–97. [Google Scholar] [CrossRef]
- Blank, L.J.; Acton, E.K.; Willis, A.W. Predictors of Mortality in Older Adults With Epilepsy: Implications for Learning Health Systems. Neurology 2021, 96, e93–e101. [Google Scholar] [CrossRef]
- Lambrecq, V.; Marchal, C.; Michel, V.; Guehl, D.; Burbaud, P.; Rougier, A. Clinical Features of Late-Onset Partial Cryptogenic Epilepsy: Toward an Idiopathic Temporal Epilepsy? Epilepsy Behav. 2013, 28, 168–171. [Google Scholar] [CrossRef]
- Green, S.F.; Loefflad, N.; Heaney, D.C.; Rajakulendran, S. New-Onset Seizures in Older People: Clinical Features, Course and Outcomes. J. Neurol. Sci. 2021, 429, 118065. [Google Scholar] [CrossRef]
- Reyes, A.; Kaestner, E.; Edmonds, E.C.; Christina Macari, A.; Wang, Z.I.; Drane, D.L.; Punia, V.; Busch, R.M.; Hermann, B.P.; McDonald, C.R.; et al. Diagnosing Cognitive Disorders in Older Adults with Epilepsy. Epilepsia 2021, 62, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Kaestner, E.; Reyes, A.; Chen, A.; Rao, J.; Macari, A.C.; Choi, J.Y.; Qiu, D.; Hewitt, K.; Wang, Z.I.; Drane, D.L.; et al. Atrophy and Cognitive Profiles in Older Adults with Temporal Lobe Epilepsy Are Similar to Mild Cognitive Impairment. Brain 2021, 144, 236–250. [Google Scholar] [CrossRef]
- Griffith, H.R.; Martin, R.C.; Bambara, J.K.; Marson, D.C.; Faught, E. Older Adults with Epilepsy Demonstrate Cognitive Impairments Compared with Patients with Amnestic Mild Cognitive Impairment. Epilepsy Behav. 2006, 8, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dejakaisaya, H.; Kwan, P.; Jones, N.C. Astrocyte and Glutamate Involvement in the Pathogenesis of Epilepsy in Alzheimer’s Disease. Epilepsia 2021, 62, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Vico Varela, E.; Etter, G.; Williams, S. Excitatory-Inhibitory Imbalance in Alzheimer’s Disease and Therapeutic Significance. Neurobiol. Dis. 2019, 127, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Heinzen, E.L.; Yoon, W.; Weale, M.E.; Sen, A.; Wood, N.W.; Burke, J.R.; Welsh-Bohmer, K.A.; Hulette, C.M.; Sisodiya, S.M.; Goldstein, D.B. Alternative Ion Channel Splicing in Mesial Temporal Lobe Epilepsy and Alzheimer’s Disease. Genome Biol. 2007, 8, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, L.; Lo, A.; Knox, K.M.; Barker-Haliski, M. Alzheimer’s Disease and Epilepsy: A Perspective on the Opportunities for Overlapping Therapeutic Innovation. Neurochem. Res. 2021, 46, 1895–1912. [Google Scholar] [CrossRef] [PubMed]
- Dulla, C.G.; Coulter, D.A.; Ziburkus, J. From Molecular Circuit Dysfunction to Disease: Case Studies in Epilepsy, Traumatic Brain Injury, and Alzheimer’s Disease. Neurosci. Rev. J. Bring. Neurobiol. Neurol. Psychiatry 2016, 22, 295–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.; Parnetti, L.; D’Amelio, M.; Tozzi, A.; Tantucci, M.; Romigi, A.; Siliquini, S.; Cavallucci, V.; Di Filippo, M.; Mazzocchetti, P.; et al. Epilepsy, Amyloid-β, and D1 Dopamine Receptors: A Possible Pathogenetic Link? Neurobiol. Aging 2016, 48, 161–171. [Google Scholar] [CrossRef]
- Sakimoto, Y.; Oo, P.M.-T.; Goshima, M.; Kanehisa, I.; Tsukada, Y.; Mitsushima, D. Significance of GABA(A) Receptor for Cognitive Function and Hippocampal Pathology. Int. J. Mol. Sci. 2021, 22, 12456. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.; Scharfman, H.E. Shared Cognitive and Behavioral Impairments in Epilepsy and Alzheimer’s Disease and Potential Underlying Mechanisms. Epilepsy Behav. 2013, 26, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicastro, N.; Assal, F.; Seeck, M. From Here to Epilepsy: The Risk of Seizure in Patients with Alzheimer’s Disease. Epileptic Disord. 2016, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.-Q.; Kreitzer, A.; et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer’s Disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Rao, N.R.; Savas, J.N. Levetiracetam Treatment Normalizes Levels of Presynaptic Endocytosis Machinery and Restores Nonamyloidogenic APP Processing in App Knock-in Mice. J. Proteome Res. 2021, 20, 3580–3589. [Google Scholar] [CrossRef]
- Putra, M.; Puttachary, S.; Liu, G.; Lee, G.; Thippeswamy, T. Fyn-Tau Ablation Modifies PTZ-Induced Seizures and Post-Seizure Hallmarks of Early Epileptogenesis. Front. Cell. Neurosci. 2020, 14, 428. [Google Scholar] [CrossRef]
- Briner, A.; Götz, J.; Polanco, J.C. Fyn Kinase Controls Tau Aggregation In Vivo. Cell Rep. 2020, 32, 108045. [Google Scholar] [CrossRef]
- Tábuas-Pereira, M.; Durães, J.; Lopes, J.; Sales, F.; Bento, C.; Duro, D.; Santiago, B.; Almeida, M.R.; Leitão, M.J.; Baldeiras, I.; et al. Increased CSF Tau Is Associated with a Higher Risk of Seizures in Patients with Alzheimer’s Disease. Epilepsy Behav. 2019, 98, 207–209. [Google Scholar] [CrossRef]
- Di Nunzio, M.; Di Sapia, R.; Sorrentino, D.; Kebede, V.; Cerovic, M.; Gullotta, G.S.; Bacigaluppi, M.; Audinat, E.; Marchi, N.; Ravizza, T.; et al. Microglia Proliferation Plays Distinct Roles in Acquired Epilepsy Depending on Disease Stages. Epilepsia 2021, 62, 1931–1945. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Qiao, P.-F.; Wan, C.-Q.; Cai, M.; Zhou, N.-K.; Li, Q. Role of Blood-Brain Barrier in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Weissberg, I.; Reichert, A.; Heinemann, U.; Friedman, A. Blood-Brain Barrier Dysfunction in Epileptogenesis of the Temporal Lobe. Epilepsy Res. Treat. 2011, 2011, 143908. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [Green Version]
- Mueed, Z.; Tandon, P.; Maurya, S.K.; Deval, R.; Kamal, M.A.; Poddar, N.K. Tau and MTOR: The Hotspots for Multifarious Diseases in Alzheimer’s Development. Front. Neurosci. 2019, 12, 1017. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Epilepsy and Cognitive Impairments in Alzheimer Disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, M.; Iadecola, C. Risk Factor for Alzheimer’s Disease Breaks the Blood-Brain Barrier. Nature 2020, 581, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.C.; Dove, G.; Cascino, G.D.; Petersen, R.C. Recurrent Seizures in Patients with Dementia: Frequency, Seizure Types, and Treatment Outcome. Epilepsy Behav. 2009, 14, 118–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and Epileptiform Activity in the Early Stages of Alzheimer Disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef]
- Belcastro, V.; Costa, C.; Galletti, F.; Pisani, F.; Calabresi, P.; Parnetti, L. Levetiracetam Monotherapy in Alzheimer Patients with Late-Onset Seizures: A Prospective Observational Study. Eur. J. Neurol. 2007, 14, 1176–1178. [Google Scholar] [CrossRef]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam Suppresses Neuronal Network Dysfunction and Reverses Synaptic and Cognitive Deficits in an Alzheimer’s Disease Model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretin, B. Treatment of Seizures in Older Patients with Dementia. Drugs Aging 2021, 38, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.-N. Treatment of Epilepsy for People with Alzheimer’s Disease. Cochrane Database Syst. Rev. 2021, 5, CD011922. [Google Scholar] [CrossRef] [PubMed]
- Cretin, B. Pharmacotherapeutic Strategies for Treating Epilepsy in Patients with Alzheimer’s Disease. Expert Opin. Pharmacother. 2018, 19, 1201–1209. [Google Scholar] [CrossRef]
- Fu, C.-H.; Iascone, D.M.; Petrof, I.; Hazra, A.; Zhang, X.; Pyfer, M.S.; Tosi, U.; Corbett, B.F.; Cai, J.; Lee, J.; et al. Early Seizure Activity Accelerates Depletion of Hippocampal Neural Stem Cells and Impairs Spatial Discrimination in an Alzheimer’s Disease Model. Cell Rep. 2019, 27, 3741–3751. [Google Scholar] [CrossRef]
- Vossel, K.; Ranasinghe, K.G.; Beagle, A.J.; La, A.; Ah Pook, K.; Castro, M.; Mizuiri, D.; Honma, S.M.; Venkateswaran, N.; Koestler, M.; et al. Effect of Levetiracetam on Cognition in Patients With Alzheimer Disease With and Without Epileptiform Activity: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 1345–1354. [Google Scholar] [CrossRef]
- Sen, A.; Akinola, M.; Tai, X.Y.; Symmonds, M.; Davis Jones, G.; Mura, S.; Galloway, J.; Hallam, A.; Chan, J.Y.C.; Koychev, I.; et al. An Investigation of Levetiracetam in Alzheimer’s Disease (ILiAD): A Double-Blind, Placebo-Controlled, Randomised Crossover Proof of Concept Study. Trials 2021, 22, 508. [Google Scholar] [CrossRef]
- Gardoni, F.; Di Luca, M. New Targets for Pharmacological Intervention in the Glutamatergic Synapse. Eur. J. Pharmacol. 2006, 545, 2–10. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Guida, M.; Vergallo, A.; Bonuccelli, U.; Zaccara, G. Treatment of Epilepsy in Patients with Alzheimer’s Disease. Expert Rev. Neurother. 2017, 17, 309–318. [Google Scholar] [CrossRef]
- Löscher, W.; Gillard, M.; Sands, Z.A.; Kaminski, R.M.; Klitgaard, H. Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond. CNS Drugs 2016, 30, 1055–1077. [Google Scholar] [CrossRef] [Green Version]
- Stockburger, C.; Miano, D.; Baeumlisberger, M.; Pallas, T.; Arrey, T.N.; Karas, M.; Friedland, K.; Müller, W.E. A Mitochondrial Role of SV2a Protein in Aging and Alzheimer’s Disease: Studies with Levetiracetam. J. Alzheimer’s Dis. 2016, 50, 201–215. [Google Scholar] [CrossRef]
- Lozupone, M.; Solfrizzi, V.; D’Urso, F.; Di Gioia, I.; Sardone, R.; Dibello, V.; Stallone, R.; Liguori, A.; Ciritella, C.; Daniele, A.; et al. Anti-Amyloid-β Protein Agents for the Treatment of Alzheimer’s Disease: An Update on Emerging Drugs. Expert Opin. Emerg. Drugs 2020, 25, 319–335. [Google Scholar] [CrossRef]
- Musaeus, C.S.; Shafi, M.M.; Santarnecchi, E.; Herman, S.T.; Press, D.Z. Levetiracetam Alters Oscillatory Connectivity in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1065–1076. [Google Scholar] [CrossRef]
- Wu, H.; Lu, M.-H.; Wang, W.; Zhang, M.-Y.; Zhu, Q.-Q.; Xia, Y.-Y.; Xu, R.-X.; Yang, Y.; Chen, L.-H.; Ma, Q.-H. Lamotrigine Reduces β-Site AβPP-Cleaving Enzyme 1 Protein Levels Through Induction of Autophagy. J. Alzheimer’s Dis. 2015, 46, 863–876. [Google Scholar] [CrossRef]
- Caccamo, D.; Pisani, L.R.; Mazzocchetti, P.; Ientile, R.; Calabresi, P.; Pisani, F.; Costa, C. Neuroprotection as a Potential Therapeutic Perspective in Neurodegenerative Diseases: Focus on Antiepileptic Drugs. Neurochem. Res. 2016, 41, 340–352. [Google Scholar] [CrossRef]
- Wang, K.; Fernandez-Escobar, A.; Han, S.; Zhu, P.; Wang, J.-H.; Sun, Y. Lamotrigine Reduces Inflammatory Response and Ameliorates Executive Function Deterioration in an Alzheimer’s-Like Mouse Model. BioMed Res. Int. 2016, 2016, 7810196. [Google Scholar] [CrossRef]
- Bang, S.R.; Ambavade, S.D.; Jagdale, P.G.; Adkar, P.P.; Waghmare, A.B.; Ambavade, P.D. Lacosamide Reduces HDAC Levels in the Brain and Improves Memory: Potential for Treatment of Alzheimer’s Disease. Pharmacol. Biochem. Behav. 2015, 134, 65–69. [Google Scholar] [CrossRef]
- Hoyt, C.T.; Domingo-Fernández, D.; Balzer, N.; Güldenpfennig, A.; Hofmann-Apitius, M. A Systematic Approach for Identifying Shared Mechanisms in Epilepsy and Its Comorbidities. Database 2018, 2018, bay050. [Google Scholar] [CrossRef] [Green Version]
- González-Sanmiguel, J.; Burgos, C.F.; Bascuñán, D.; Fernández-Pérez, E.J.; Riffo-Lepe, N.; Boopathi, S.; Fernández-Pérez, A.; Bobadilla-Azócar, C.; González, W.; Figueroa, M.; et al. Gabapentin Inhibits Multiple Steps in the Amyloid Beta Toxicity Cascade. ACS Chem. Neurosci. 2020, 11, 3064–3076. [Google Scholar] [CrossRef]
- Supasitthumrong, T.; Bolea-Alamanac, B.M.; Asmer, S.; Woo, V.L.; Abdool, P.S.; Davies, S.J.C. Gabapentin and Pregabalin to Treat Aggressivity in Dementia: A Systematic Review and Illustrative Case Report. Br. J. Clin. Pharmacol. 2019, 85, 690–703. [Google Scholar] [CrossRef]
- Long, Z.-M.; Zhao, L.; Jiang, R.; Wang, K.-J.; Luo, S.-F.; Zheng, M.; Li, X.-F.; He, G.-Q. Valproic Acid Modifies Synaptic Structure and Accelerates Neurite Outgrowth Via the Glycogen Synthase Kinase-3β Signaling Pathway in an Alzheimer’s Disease Model. CNS Neurosci. Ther. 2015, 21, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Loy, R.; Tariot, P.N. Neuroprotective Properties of Valproate: Potential Benefit for AD and Tauopathies. J. Mol. Neurosci. 2002, 19, 303–307. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Li, X.-J.; Zhang, H.-Y. Valproic Acid as a Promising Agent to Combat Alzheimer’s Disease. Brain Res. Bull. 2010, 81, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Seibert, M.; Mühlbauer, V.; Holbrook, J.; Voigt-Radloff, S.; Brefka, S.; Dallmeier, D.; Denkinger, M.; Schönfeldt-Lecuona, C.; Klöppel, S.; von Arnim, C.A.F. Efficacy and Safety of Pharmacotherapy for Alzheimer’s Disease and for Behavioural and Psychological Symptoms of Dementia in Older Patients with Moderate and Severe Functional Impairments: A Systematic Review of Controlled Trials. Alzheimer’s Res. Ther. 2021, 13, 131. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Suggested Risk Factors of Epilepsy in AD |
|---|
| Sociodemographic: Male sex. Younger age at the onset of symptoms (both in sporadic and autosomal dominant AD). Clinical and anatomic features: Longer disease duration (in years). Disease severity. Neuroimaging features: greater affectation of precuneus and atrophy pattern with parietal predominance. Chronic use of drugs that reduce seizure threshold (for instance, classic antipsychotics). Comorbidities increasing the risk of epilepsy: Cerebrovascular pathology (micro- and macrovascular damage, mainly with when there is cortical involvement). Brain traumatic injury. |
| SV2A Ligands | Na+ Channel Blockers | Multiple Mechanisms | Ca+ Channel Blockers | AMPAR Blocker | |||||
|---|---|---|---|---|---|---|---|---|---|
| Levetiracetam (LEV) | Brivaracetam (BVT) | Lamotrigine (LTG) | Lacosamide (LCS) | “Zepines” (CBZ, OXC, ESL) | Valproic Acid (VPA) | Zonisamide (ZNS) and Topiramate (TPM) | Pregabalin (PGB) and Gabapentin (GBP) | Perampanel (PER) | |
| Mechanism of action | - Binds SV2A. - Blocks AMPA and NMDAR (reduces release of glutamate). - Induces GABA potentiation. - Effect on glycine or kainic-acid currents. | - Binds SV2A (20-fold higher affinity compared to LEV). -Minor block on NMDAR. | - Blocks voltage-dependent sodium channels. | - Blocks voltage-dependent sodium channels (enhancing slow inactivation). | - Blocks voltage-dependent sodium channels. | - GABA potentiation. - Blocks T-type calcium channels, sodium channels, and NMDAR. | - GABA potentiation (only TPM). - Blocks AMPAR (only TPM), T-type calcium channels (only ZNS), and voltage-dependent sodium channels. | - Blocks voltage-dependent calcium channels. | - AMPA glutamate receptor antagonist. |
| Spectrum of efficacy | - Broad-spectrum. Including antimyoclonic effect. | - Focal seizures. - Preclinical models: broad-spectrum efficacy. | - Broad-spectrum. | - Focal seizures. | - Focal seizures. | - Broad-spectrum. | - Broad-spectrum. | - Focal seizures. | - Focal seizures, generalized seizures (only as adjunctive therapy), useful for myoclonic seizures. |
| Clinical experience in AD | - First-line treatment. - Safety and absence of interactions. | - Well tolerated. - Less irritability than LEV. - Alternative for LEV or LTG. | - First-line treatment. - Less sedative and few cognitive adverse effects. | - Well tolerated. - Alternative for LEV or LTG. | - Not considered as first- or second-line treatment. | - Not considered as first- or second-line treatment. | - Not considered as first- or second-line treatment. | - Not considered as first- or second-line treatment. | - Possible alternative treatment, study data are lacking. - No data on cognitive side effects. |
| Potential limitations and risks in AD | - Dose-dependent somnolence and irritability. - 10–15% stop due to neuropsychiatric side effects. | - Irritability but with lower frequency compared to LEV. | - Unsteadiness. - Onset insomnia. - May exacerbate myoclonic seizures. | - Unsteadiness (less frequent than others Na+ blockers). - May exacerbate myoclonic seizures. | - Cognitive impairment related with decreased cholinergic tone (less frequent with ESL). - Unsteadiness. | - Encephalopathy, hyperammonemia. - May induce cognitive impairment and/or motor worsening (tremor). | - Cognitive adverse effects (less frequent with ZNS). | - Less effective. - Cognitive slowing. - Dizziness. | - Dizziness. - Aggression and hostility (special caution if neuropsychiatric symptoms with LEV). |
| Neutral. | Acetylcholinesterase inhibitors. |
| Antidepressants: Selective serotonin reuptake inhibitors. | |
| Antipsychotics: Quetiapine and risperidone. | |
| Decrease seizure threshold. | Antidepressants a: Tricyclic antidepressants and bupropion. |
| Antipsychotics b: Clozapine, chlorpromazine and haloperidol. | |
| Controversy. | Memantine c |
| SV2A Ligands | Na+ Channel Blockers | Multiple Mechanisms | Ca+ Channel Blockers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Levetiracetam (LEV) | Brivaracetam (BVT) | Lamotrigine (LTG) | Lacosamide (LCS) | “Zepines” (CBZ, OXC, ESL) | Valproic Acid (VPA) | Zonisamide (ZNS) and Topiramate (TPM) | Pregabalin (PGB) and Gabapentin (GBP) | ||
| HUMAN MODELS | - Improve attention, verbal fluency, visuospatial functions, and hippocampal-related memory tasks. - Reduce hippocampal hyperactivity (assessed by fMRI and EEG). | - Expected to be similar to LEV. | - Better performance in naming and recognition tasks. - Improvement of affective symptoms (mainly depression). | - Single study in LOEU: improve verbal fluency but no other cognitive domains. | |||||
| ANIMAL MODELS AND CELL CULTURES | Fibrillar and amyloid plaque deposition | - ↓ Aβ42 oligomers and fibrils, and amyloid plaque burden. | - ↓ BACE1 (via ↓mTOR): ↓ amyloid plaque density | - ↓ Aβ plaques | - ↓ Aβ oligomers and formation of neuritic plaques. | - Neuro-protection: interfere with Aβ-induced toxicity. | |||
| Tau deposition and/or hyperphosphorylation | - ↓ Aβ-induced hyperphosphorylation of tau. | - ↓ GSK3β activity: ↓ p-tau. | - ↓ GSK3β: ↓ p-tau. | ||||||
| Neurogenesis and/or hippocampal remodeling | - Modify positively hippocampal remodeling. - Restore neurogenesis. | - ↓ CA1 hippocampal neuronal loss. - ↓ HDAC, ↑BCL2: neurogenesis in the granule cell layer of dentate gyrus. | - ↓ HDAC activity. | - ↑ bcl-2:↓ apoptosis. - ↑ Neuronal progenitor proliferation by ↑ cyclin D2. | - ↓ HDAC activity. | ||||
| Others | - Repair mitochondrial dysfunction. - Modify the excitotoxicity mediated by GLUT. - Improve synaptic function. - ↓ hippocampal hyperexcitability. | - Normalize the E-I system imbalance. - Modify sensitivity of synaptic vesicles to Ca+: reduce release of NT (GLUT and GABA) in hippocampus. | - ↓ Neuroinflammation - ↓ GLUT release. | - ↓ GLUT-mediated excitatory signaling. - ↑ NA tone. | - ↑ GABAergic neuron differentiation. - ↓Neuroinflammation. | - ↑ GABAergic tone. | - ↓ neuronal hyperexcitability. | ||
| Cognitive function improvement | - Improve learning and memory deficits and spatial discrimination tasks. | - Enhance performance in memory tasks. | - Ameliorate executive dysfunction. | - May improve disrupted memory. | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altuna, M.; Olmedo-Saura, G.; Carmona-Iragui, M.; Fortea, J. Mechanisms Involved in Epileptogenesis in Alzheimer’s Disease and Their Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 4307. https://doi.org/10.3390/ijms23084307
Altuna M, Olmedo-Saura G, Carmona-Iragui M, Fortea J. Mechanisms Involved in Epileptogenesis in Alzheimer’s Disease and Their Therapeutic Implications. International Journal of Molecular Sciences. 2022; 23(8):4307. https://doi.org/10.3390/ijms23084307
Chicago/Turabian StyleAltuna, Miren, Gonzalo Olmedo-Saura, María Carmona-Iragui, and Juan Fortea. 2022. "Mechanisms Involved in Epileptogenesis in Alzheimer’s Disease and Their Therapeutic Implications" International Journal of Molecular Sciences 23, no. 8: 4307. https://doi.org/10.3390/ijms23084307
APA StyleAltuna, M., Olmedo-Saura, G., Carmona-Iragui, M., & Fortea, J. (2022). Mechanisms Involved in Epileptogenesis in Alzheimer’s Disease and Their Therapeutic Implications. International Journal of Molecular Sciences, 23(8), 4307. https://doi.org/10.3390/ijms23084307