
3-Methylglutaconic Aciduria Type I Due to AUH Defect: The Case Report of a Diagnostic Odyssey and a Review of the Literature

 , ,
, ,
Abstract
:1. Introduction
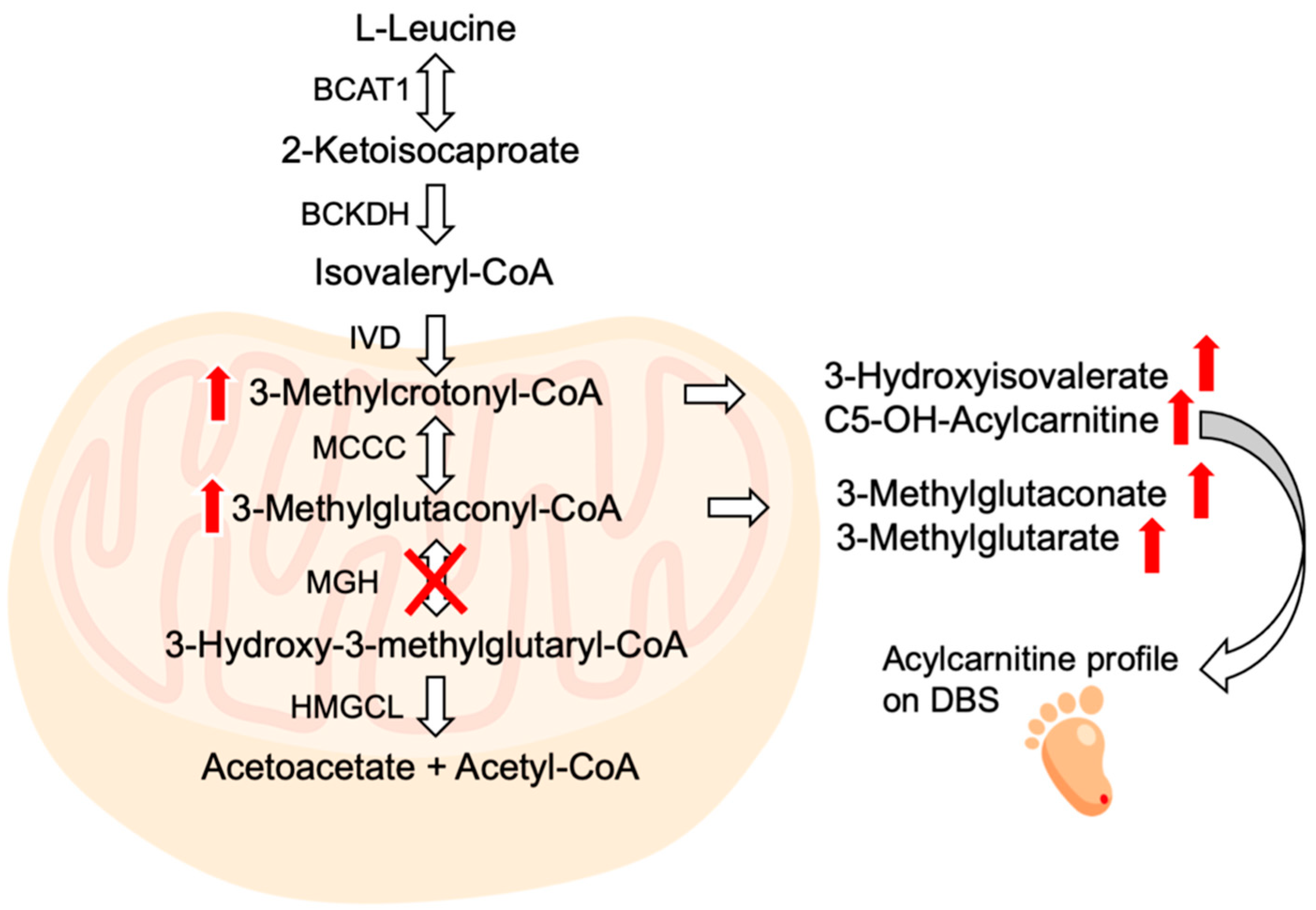
2. Results
2.1. Case Report
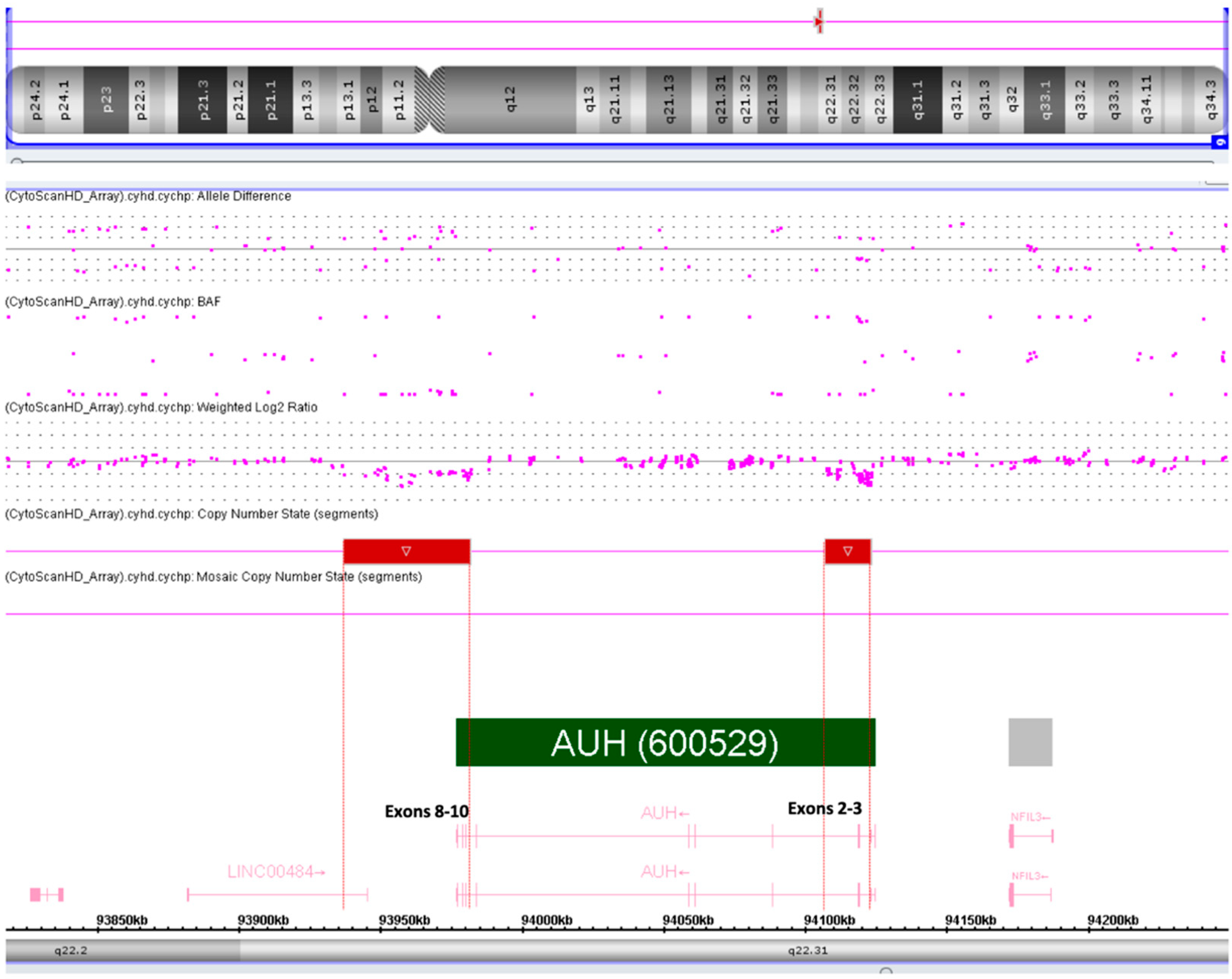
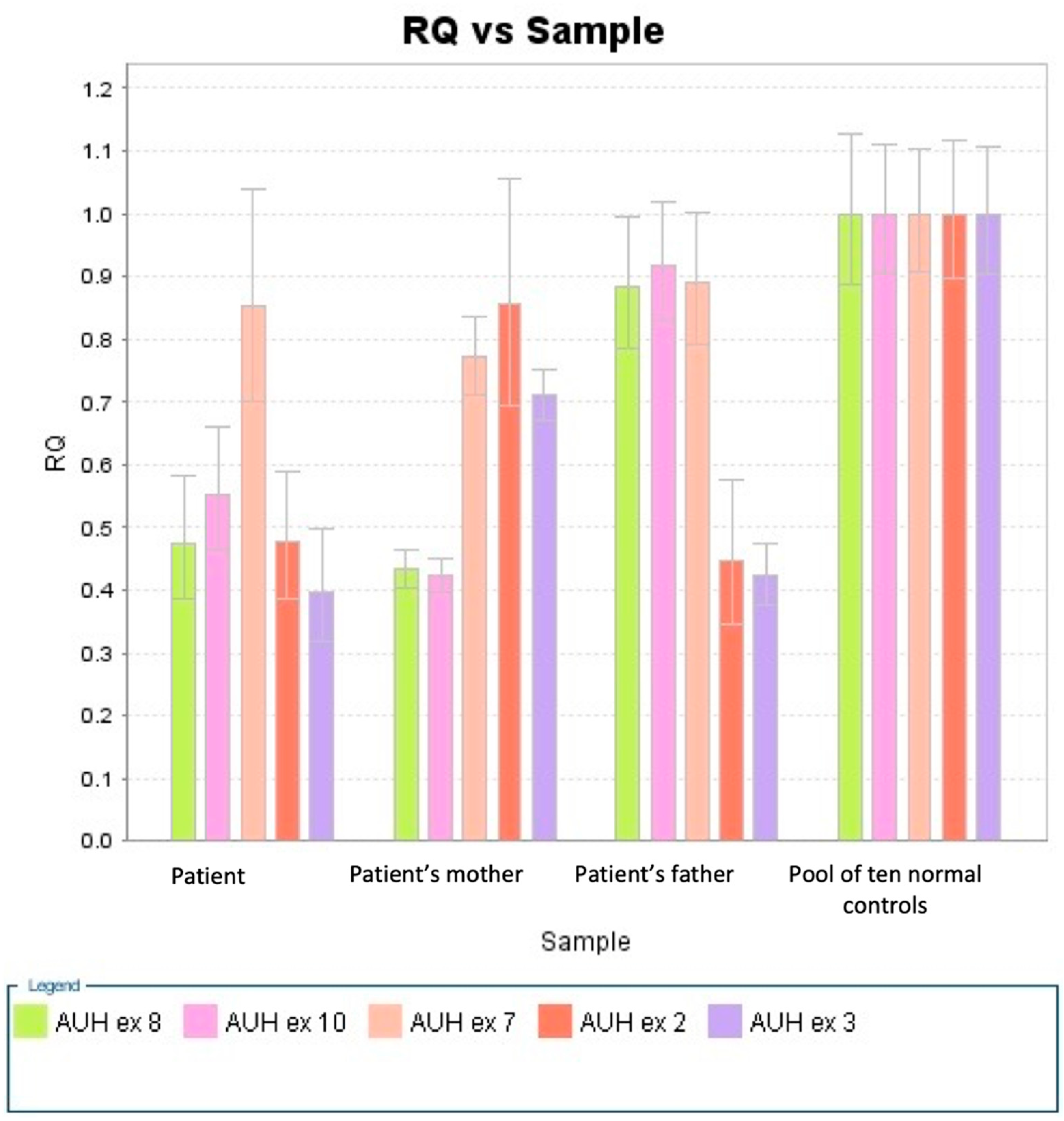
2.2. Diagnostic Procedures
2.3. Clinical Follow-Up
3. Discussion
4. Materials and Methods
4.1. Case Report
4.2. NGS
4.3. SNP Array Analysis
4.4. CNV Assays
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- IJlst, L.; Loupatty, F.J.; Ruiter, J.P.N.; Duran, M.; Lehnert, W.; Wanders, R.J.A. 3-Methylglutaconic Aciduria Type I Is Caused by Mutations in AUH. Am. J. Hum. Genet. 2002, 71, 1463–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, M.; Schniegler-Mattox, U.; Peters, V.; Hoffmann, G.F.; Liesert, M.; Buckel, W.; Zschocke, J. Biochemical Characterization of Human 3-Methylglutaconyl-CoA Hydratase and Its Role in Leucine Metabolism: Human 3-Methylglutaconyl-CoA Hydratase. FEBS J. 2006, 273, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.B.N.; Peters, V.; Gibson, K.M.; Liesert, M.; Buckel, W.; Wilcken, B.; Carpenter, K.; Ensenauer, R.; Hoffmann, G.F.; Mack, M.; et al. Mutations in TheAUH Gene Cause 3-Methylglutaconic Aciduria Type I. Hum. Mutat. 2003, 21, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.R.; Saudubray, J.-M.; Walter, J.H. (Eds.) Inborn Metabolic Diseases: Diagnosis and Treatment, 6th ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Duran, M.; Beemer, F.A.; Tibosch, A.S.; Bruinvis, L.; Ketting, D.; Wadman, S.K. Inherited 3-Methylglutaconic Aciduria in Two Brothers--Another Defect of Leucine Metabolism. J. Pediatr. 1982, 101, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Narisawa, K.; Gibson, K.M.; Sweetman, L.; Nyhan, W.L.; Duran, M.; Wadman, S.K. Deficiency of 3-Methylglutaconyl-Coenzyme A Hydratase in Two Siblings with 3-Methylglutaconic Aciduria. J. Clin. Investig. 1986, 77, 1148–1152. [Google Scholar] [CrossRef] [Green Version]
- Kirkby, B.; Roman, N.; Kobe, B.; Kellie, S.; Forwood, J.K. Functional and Structural Properties of Mammalian Acyl-Coenzyme A Thioesterases. Prog. Lipid Res. 2010, 49, 366–377. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Tucker, T.; Casey, B. Phenotypic Heterogeneity in Two Siblings with 3-Methylglutaconic Aciduria Type I Caused by a Novel Intragenic Deletion. Mol. Genet. Metab. 2011, 104, 410–413. [Google Scholar] [CrossRef]
- Tavasoli, A.R.; Shervin Badv, R.; Zschocke, J.; Ashrafi, M.R.; Rostami, P. Early Infantile Presentation of 3-Methylglutaconic Aciduria Type 1 with a Novel Mutation in AUH Gene: A Case Report and Literature Review. Brain Dev. 2017, 39, 714–716. [Google Scholar] [CrossRef]
- Gibson, K.M.; Wappner, R.S.; Jooste, S.; Erasmus, E.; Mienie, L.J.; Gerlo, E.; Desprechins, B.; De Meirleir, L. Variable Clinical Presentation in Three Patients with 3-Methylglutaconyl-Coenzyme A Hydratase Deficiency. J. Inherit. Metab. Dis. 1998, 21, 631–638. [Google Scholar] [CrossRef]
- Ensenauer, R.; Müller, C.B.; Schwab, K.O.; Gibson, K.M.; Brandis, M.; Lehnert, W. 3-Methylglutaconyl-CoA Hydratase Deficiency: A New Patient with Speech Retardation as the Leading Sign. J. Inherit. Metab. Dis. 2000, 23, 341–344. [Google Scholar] [CrossRef]
- Gibson, K.M.; Lee, C.F.; Wappner, R.S. 3-Methylglutaconyl-Coenzyme-A Hydratase Deficiency: A New Case. J. Inherit. Metab. Dis. 1992, 15, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Jooste, S.; Erasmus, E.; Mienie, L.J.; de Wet, W.J.; Gibson, K.M. The Detection of 3-Methylglutarylcarnitine and a New Dicarboxylic Conjugate, 3-Methylglutaconylcarnitine, in 3-Methylglutaconic Aciduria. Clin. Chim. Acta 1994, 230, 1–8. [Google Scholar] [CrossRef]
- Dudipala, S.C.; Prashanthi, M.; Chenalla, L.K. Acute Encephalopathic Presentation of 3-Methylglutaconic Aciduria Type I With a Novel Mutation in AUH Gene. Cureus 2020, 12, e11951. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Takahashi, T.; Sawaishi, Y.; Ishida, A.; Matsumori, M.; Shoji, Y.; Enoki, M.; Watanabe, H.; Takada, G. 3-Methylglutaconic Aciduria Type I: Clinical Heterogeneity as a Neurometabolic Disease. J. Inherit. Metab. Dis. 1999, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, M.; Shoji, Y.; Takahashi, T.; Shoji, Y.; Takada, G. A Molecular Lesion in a Japanese Patient with Severe Phenotype of 3-Methylglutaconic Aciduria Type I. Pediatr. Int. 2005, 47, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Illsinger, S.; Lücke, T.; Zschocke, J.; Gibson, K.M.; Das, A.M. 3-Methylglutaconic Aciduria Type I in a Boy with Fever-Associated Seizures. Pediatr. Neurol. 2004, 30, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Engelke, U.F.H.; Kremer, B.; Kluijtmans, L.A.J.; van der Graaf, M.; Morava, E.; Loupatty, F.J.; Wanders, R.J.A.; Moskau, D.; Loss, S.; van den Bergh, E.; et al. NMR Spectroscopic Studies on the Late Onset Form of 3-Methylglutaconic Aciduria Type I and Other Defects in Leucine Metabolism. NMR Biomed. 2006, 19, 271–278. [Google Scholar] [CrossRef]
- Eriguchi, M.; Mizuta, H.; Kurohara, K.; Kosugi, M.; Yakushiji, Y.; Okada, R.; Yukitake, M.; Hasegawa, Y.; Yamaguchi, S.; Kuroda, Y. 3-Methylglutaconic Aciduria Type I Causes Leukoencephalopathy of Adult Onset. Neurology 2006, 67, 1895–1896. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Kremer, B.H.; Graham, A.; Willemsen, M.A.; Loupatty, F.J.; Hogg, S.L.; Engelke, U.F.; Kluijtmans, L.A.; Wanders, R.J.; Illsinger, S.; et al. 3-Methylglutaconic Aciduria Type I Redefined: A Syndrome with Late-Onset Leukoencephalopathy. Neurology 2010, 75, 1079–1083. [Google Scholar] [CrossRef]
- Spergel, C.D.; Milko, M.; Edwards, C.; Steinhoff, J.P. 3-Methylglutaconyl-Coenzyme-A Hydratase Deficiency and the Development of Dilated Cardiomyopathy. Cardiol. Res. 2014, 5, 158–162. [Google Scholar] [CrossRef] [Green Version]
- Bizjak, N.; Zerjav Tansek, M.; Avbelj Stefanija, M.; Repic Lampret, B.; Mezek, A.; Drole Torkar, A.; Battelino, T.; Groselj, U. Precocious Puberty in a Girl with 3-Methylglutaconic Aciduria Type 1 (3-MGA-I) Due to a Novel AUH Gene Mutation. Mol. Genet. Metab. Rep. 2020, 25, 100691. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jungbluth, H.; Sewry, C.A.; Feng, L.; Bertini, E.; Bushby, K.; Straub, V.; Roper, H.; Rose, M.R.; Brockington, M.; et al. Molecular Mechanisms and Phenotypic Variation in RYR1-Related Congenital Myopathies. Brain 2007, 130, 2024–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Brockington, M.; Jungbluth, H.; Monk, D.; Stanier, P.; Sewry, C.A.; Moore, G.E.; Muntoni, F. Epigenetic Allele Silencing Unveils Recessive RYR1 Mutations in Core Myopathies. Am. J. Hum. Genet. 2006, 79, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, S.B.; Duran, M.; Anikster, Y.; Barth, P.G.; Sperl, W.; Zschocke, J.; Morava, E.; Wevers, R.A. Inborn Errors of Metabolism with 3-Methylglutaconic Aciduria as Discriminative Feature: Proper Classification and Nomenclature. J. Inherit. Metab. Dis. 2013, 36, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Kluijtmans, L.A.J.; Rodenburg, R.J.; Sass, J.O.; Nouws, J.; van Kaauwen, E.P.; Kleefstra, T.; Tranebjaerg, L.; de Vries, M.C.; Isohanni, P.; et al. 3-Methylglutaconic Aciduria—Lessons from 50 Genes and 977 Patients. J. Inherit. Metab. Dis. 2013, 36, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Vamecq, J.; Papegay, B.; Nuyens, V.; Boogaerts, J.; Leo, O.; Kruys, V. Mitochondrial Dysfunction, AMPK Activation and Peroxisomal Metabolism: A Coherent Scenario for Non-Canonical 3-Methylglutaconic Acidurias. Biochimie 2020, 168, 53–82. [Google Scholar] [CrossRef]
- Gibson, K.M.; Breuer, J.; Kaiser, K.; Nyhan, W.L.; McCoy, E.E.; Ferreira, P.; Greene, C.L.; Blitzer, M.G.; Shapira, E.; Reverte, F. 3-Hydroxy-3-Methylglutaryl-Coenzyme A Lyase Deficiency: Report of Five New Patients. J. Inherit. Metab. Dis. 1988, 11, 76–87. [Google Scholar] [CrossRef]
- Grünert, S.C.; Schlatter, S.M.; Schmitt, R.N.; Gemperle-Britschgi, C.; Mrázová, L.; Balcı, M.C.; Bischof, F.; Çoker, M.; Das, A.M.; Demirkol, M.; et al. 3-Hydroxy-3-Methylglutaryl-Coenzyme A Lyase Deficiency: Clinical Presentation and Outcome in a Series of 37 Patients. Mol. Genet. Metab. 2017, 121, 206–215. [Google Scholar] [CrossRef]
- Coelho, M.P.; Correia, J.; Dias, A.; Nogueira, C.; Bandeira, A.; Martins, E.; Vilarinho, L. Iron-sulfur Cluster ISD11 Deficiency (LYRM4 Gene) Presenting as Cardiorespiratory Arrest and 3-methylglutaconic Aciduria. JIMD Rep. 2019, 49, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Oláhová, M.; Yoon, W.H.; Thompson, K.; Jangam, S.; Fernandez, L.; Davidson, J.M.; Kyle, J.E.; Grove, M.E.; Fisk, D.G.; Kohler, J.N.; et al. Biallelic Mutations in ATP5F1D, Which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder. Am. J. Hum. Genet. 2018, 102, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Huffnagel, I.C.; Redeker, E.J.W.; Reneman, L.; Vaz, F.M.; Ferdinandusse, S.; Poll-The, B.T. Mitochondrial Encephalopathy and Transient 3-Methylglutaconic Aciduria in ECHS1 Deficiency: Long-Term Follow-Up. In JIMD Reports; JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 39, pp. 83–87. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R.; American College of Medical Genetics Newborn Screening Expert Group. Newborn Screening: Toward a Uniform Screening Panel and System—Executive Summary. Pediatrics 2006, 117 (Suppl. S3), S296–S307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical Standards for the Interpretation and Reporting of Constitutional Copy-Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caciotti, A.; Tonin, R.; Rigoldi, M.; Ferri, L.; Catarzi, S.; Cavicchi, C.; Procopio, E.; Donati, M.A.; Ficcadenti, A.; Fiumara, A.; et al. Optimizing the Molecular Diagnosis of GALNS: Novel Methods to Define and Characterize Morquio-A Syndrome-Associated Mutations. Hum. Mutat. 2015, 36, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| References | Ancestry (Consanguinity) | AUH Variant | MGH Activity (r.v.) | Urinary 3-MGA (r.v.) | Urinary 3-HIVA (r.v.) | Neuroimaging/ Age at | MRI: WM Changes | MRI: Additional Findings |
|---|---|---|---|---|---|---|---|---|
| ID 1 [3,5,6,20] | Moroccan (-) | c.589C > T p.(Arg197*)/c.589C > T p.(Arg197*) | F: 11 ± 4 pmol/min per mg protein (495 ± 89) | 519–840 mmol/mol creat (<5.66) | 144–206 mmol/mol creat (<8.15) | NA | NA | NA |
| ID 2 [1,3,5,6,20] | Moroccan (-) | c.589C > T p.(Arg197*)/c.589C > T p.(Arg197*) | F: < 0.1 nmol/min/mg protein (2.8 ± 0.6) | 762–930 mmol/mol creat (<5.66) | 172–264 mmol/mol creat (<8.15) | NA | NA | NA |
| ID 3 [12] | NA (NA) | NA | F: 3-HB/CoA esters 0.5 (2–63) L: 3-HB/CoA esters 1–3 (7–20) * | Marked elevation | Marked elevation | CT normal | NA | NA |
| ID 4 [3,10,13,20] | NA (NA) | c.719C > T p.(Ala240Val)/c.613dupA p.(Met205Asnfs*5) | F: 3-HB/CoA esters 0.2–0.55 (5.3 ± 1.2) * | 249–953 mmol/mol creat (<9) | 47–443 mmol/mol creat (<46) | CT normal | NA | NA |
| ID 5 [10] | White (NA) | NA | F: 3-HB/CoA esters 0.16–0.35 (5.3 ± 1.2) * | Marked elevation | Marked elevation | NA | NA | NA |
| ID 6 [10] | NA (+) | NA | F: 3-HB/CoA esters 0.16–0.58 (5.3 ± 1.2) * | 482–1153 mmol/mol creat (<9) | 374–3840 mmol/mol creat (<46) | MRI | Abnormal signals in the white matter | Globus pallidum hyperintensity images, cavum septum pellucidum, and cavum vergae |
| ID 7 [15,16,20] | Japanese (+) | c.263-2A > G/c.263-2A > G | F: 3-HB/CoA esters 0.3 (5.0–10.6) * L: 3-HB/CoA esters 0.2 (2.3–5.7) * | 168 mmol/mol creat (0–15) | 292 mmol/mol creat (0–4) | MRI/9 months and 23 months | Myelination almost normal | Cerebral atrophy and progressive basal ganglia atrophy |
| ID 8 [1,3,11,20] | Afghan (+) | c.895-1G > A/c.895-1G > A | F: < 0.1 nmol/min/mg protein (2.8 ± 0.6) | 400 mmol/mol creat (<9) | Increased | MRI/paediatric | NR | NR |
| ID 9 [3,20] | Lebanese (+) | c.80delG p.(Ser27Metfs*8)/c.80delG p.(Ser27Metfs*8) | F: 3-HB/CoA esters 0.06 (1.2–4.5) * | 1500–2900 mmol/mol creat | No increase | NA | NA | NA |
| ID 10 [17,20] | German (-) | c.943-2A > G/c.943-2A > G | F: markedly decreased | 570 mmol/mol creat (<9) | 500 mmol/mol creat (<67) | MRI/10 years | Mild abnormalities in deep frontal WM sparing the corpus callosum and the U-fibers | NR |
| ID 11 [18,20] | Dutch (-) | c.559G > A p.(Gly187Ser)/c.650G > A p.(Gly217Asp) | F: undetectable (2.1 ± 0.7 nmol/min/mg protein) | 94–141 mmol/mol creat (1.0–6.5) # | 61–63 mmol/mol creat (3–15) | MRI/61 years | Confluent lesions of deep and subcortical WM, sparing the corpus callosum and periventricular rim | Cranial MRS: 3-HIVA peak |
| ID 12 [19,20] | NA (+) | c.895-1G > A/c.895-1G > A | NA | 108 mmol/mol creat (<4.2) | NA | MRI/adult | WM hyperintensity extending into the subcortical U-fibers and middle cerebellar peduncle | NR |
| ID 13 [20] | British (+) | c.991A > T p.(Lys331*)/c.991A > T p.(Lys331*) | F/L: undetectable | 78 mmol/mol creat | Mildly elevated | MRI/50 years | Confluent lesions of deep and subcortical WM, sparing the corpus callosum and periventricular rim and parietooccipital regions | NR |
| ID 14 [8] | Pakistani (+) | Homozygous deletion of exons 1–3 | NA | 174.24 mmol/mol creat (<12.42) | 88.7 mmol/mol creat (<37.7) | MRI | Bilateral patchy hyperintensity in the frontal and parietal subcortical WM | Cranial MRS: 3-HIVA peak |
| ID 15 [8] | Pakistani (+) | Homozygous deletion of exons 1–3 | F: undetectable (7.7 ± 1.4 nmol/min mg) | 261.4 mmol/mol creat (<12.42) | 242.6 mmol/mol creat (<37.7) | NA | NA | NA |
| ID 16 [21] | Canadian (NA) | NA | F: reduced | Increased | NA | NA | NA | NA |
| ID 17 [9] | NA (+) | c.179delG p.(Gly60Valfs*12)/c.179delG p.(Gly60Valfs*12) | NA | Increased | Increased | MRI/2 years and 3.5 years | Delayed myelination in the trigone region, a few nonspecific hyperintensities in the centrum semiovale improved at the second study (treatment?) | NR |
| ID 18 [14] | NA (+) | c.505 + 1G > C/c.505 + 1G > C | NA | Increased | Increased | MRI/3 years | NR | NR |
| ID 19 [22] | Caucasian (-) | c.330 + 5G > A/c.330 + 5G > A | L: 0.02 nmol/min/mg protein (1.4–4.6) | Increased | Increased | MRI/6, 9, and 10 years | Progressive hyperintensive lesions in the centrum semiovale, bilateral subcortical frontal WM, and the deep frontoparietal WM | NR |
| ID 20 | Italian (-) | L: <0.02 nmol/min/mg protein (1.4–4.6) | Marked elevation | Marked elevation | NA | NA | NA |
| References | Age at Onset/ Gender | Age at Last Examination | Decompensation/ Acute Encephalopathy | Neurodevelopmental Disorders | Neurological Phenotype | Other Clinical Features | Treatment | ||
|---|---|---|---|---|---|---|---|---|---|
| Levocarnitine (Dosage) | Leucine/Protein Restricted Diet | Outcome after/Effect of Treatment | |||||||
| ID 1 [3,5,6,20] | 7 years/M—sib | 7 years | One attack of unconsciousness that lasted for almost a day. Fasting hypoglycaemia | GDD | Normal | Nocturnal enuresis | NR | NR | NR |
| ID 2 [1,3,5,6,20] | -/M—sib | 5.4 years | NR | Speech disorder | Normal | NR | NR | NR | NR |
| ID 3 [12] | 4 months/M | NA | One episode of decompensation during viral illness | NR | Normal | Bronchiolitis and gastro-oesophageal reflux | Yes (unknown) | Leucine intake 100–120 mg/kg/day | Normalization of metabolic alterations |
| ID 4 [3,10,13,20] | Neonatal/F | 13 years | Vomiting after birth. Relapsing encephalopathy during upper respiratory infections | NR | Insomnia, irritability after birth, persistent crying fits, self-mutilation | Hepatomegaly | Yes (unknown) | Yes | Ineffective leucine-restricted diet. L-carnitine: improvement of clinical (no feeding difficulties and insomnia, hepatomegaly cleared) and metabolic alterations |
| ID 5 [10] | Neonatal/M | NA | Severe acidosis, foetal distress with hypoxic ischaemic encephalopathy | Moderate GDD | Severe dystonic cerebral palsy with generalized hyperreflexia and irritability | Gastro-oesophageal reflux | Yes (unknown) | Low-protein diet using natural foods (1 g/kg/day) | No more episodes of vomiting, lethargy, and acidosis nor developmental plateau or regression |
| ID 6 [10] | -/M | 4 years | Seizures possibly associated with episodes of hypoglycaemia | Severe GDD | Severe hypotonia and intermittent thrusting in the sitting position, seizures | Premature birth, frequent upper respiratory infections, lumbar scoliosis, hepatomegaly | Yes (unknown) | NR | NA |
| ID 7 [15,16,20] | 4 months/M | 43 months | Persistent metabolic acidosis | Profound GDD | Spastic quadriplegia, athetoid dystonic movements of upper limbs | Failure to thrive | NR | Protein-restricted diet (1.0 g/kg per day; L-leucine 80 mg/kg per day) using a leucine-free formula | Improvement of metabolic alteration, marginal clinical improvement (reacquisition of eye-pursuits and social smiles) |
| ID 8 [1,3,11,20] | 1 year/M | 4 years | NR | GDD 4y: IQ 83, sustained attention deficit | Normal | Primary nocturnal and diurnal enuresis | NR | Restricted protein intake (1.5 g/kg body weight per day) | Clinical improvement |
| ID 9 [3,20] | NBS/M—sib | 9 years | NR | NR | Normal | NR | NR | NR | NR |
| ID 10 [17,20] | 1 year/M | 10 years | NR | Attention-deficit/hyperactivity disorder | Febrile seizures (15 up to age 7) | NR | NR | NR | Mild improvement of seizure severity with emergency protocol |
| ID 11 [18,20] | 35 years/F | 61 years | NR | NR | Progressive spastic ataxia | NR | NR | NR | NR |
| ID 12 [19,20] | 52 years/F | 55 years | NR | NR | Dementia, spasticity, ataxia | Urinary incontinence at age 52 | NR | NR | NR |
| ID 13 [20] | 30 years/M | 59 years | NR | NR | Progressive spastic ataxia and dementia | NR | NR | NR | NR |
| ID 14 [8] | 10 years/F—sib | 14 years | NR | IQ 82 at 10 years, arithmetic and learning disability, attention deficit | Mild uncoordination | NR | 50 mg/kg/day | Modified protein intake (1 g/kg/day natural protein intake) | Stable course |
| ID 15 [8] | 3 years/M—sib | 9 years | NR | Severe expressive language delay at 3 years | Dysarthria, two febrile seizures | History of dislike of meat | 50 mg/kg/day | Modified protein intake (1 g/kg/day natural protein intake) | Stable course |
| ID 16 [21] | 1 year/M | 25 years | Encephalopathy with metabolic decompensation during sepsis | Motor deficit, developmental learning delays | Cyanotic breath-holding spells, seizures at 1 year | Renal and heart failure during sepsis at 25 y, dilated cardiomyopathy | NR | NR | NR |
| ID 17 [9] | 22 months/M | 3.5 years | NR | Delayed milestone achievements with near normal cognition | Central hypotonia, intention tremor | Moderate sensorineural hearing loss | 100 mg/kg/day in three doses | Restricted leucine diet (60 mg/kg/day) | Incoordination, neurocognitive improvement |
| ID 18 [14] | 3 years/F | 5 years | Status dystonicus during febrile illness, irritability, unable to sleep | NR | Normal | NR | NR | Low-leucine diet | No further dystonic relapses after two years |
| ID 19 [22] | 4.5 years/F | 11 years | NR | Learning disability, attention deficit | Central hypotonia, intention tremor, and dysdiadochokinesia | Central precocious puberty | 85 mg/kg/day | 60 mg/kg/day | Improvement of attention |
| ID 20 | NBS/F | 31 months | NR | Expressive language disorder | Clumsiness | NR | 80 mg/kg/day | Slow decrease in protein intake | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardecchia, F.; Caciotti, A.; Giovanniello, T.; De Leo, S.; Ferri, L.; Galosi, S.; Santagata, S.; Torres, B.; Bernardini, L.; Carducci, C.; et al. 3-Methylglutaconic Aciduria Type I Due to AUH Defect: The Case Report of a Diagnostic Odyssey and a Review of the Literature. Int. J. Mol. Sci. 2022, 23, 4422. https://doi.org/10.3390/ijms23084422
Nardecchia F, Caciotti A, Giovanniello T, De Leo S, Ferri L, Galosi S, Santagata S, Torres B, Bernardini L, Carducci C, et al. 3-Methylglutaconic Aciduria Type I Due to AUH Defect: The Case Report of a Diagnostic Odyssey and a Review of the Literature. International Journal of Molecular Sciences. 2022; 23(8):4422. https://doi.org/10.3390/ijms23084422
Chicago/Turabian StyleNardecchia, Francesca, Anna Caciotti, Teresa Giovanniello, Sabrina De Leo, Lorenzo Ferri, Serena Galosi, Silvia Santagata, Barbara Torres, Laura Bernardini, Claudia Carducci, and et al. 2022. "3-Methylglutaconic Aciduria Type I Due to AUH Defect: The Case Report of a Diagnostic Odyssey and a Review of the Literature" International Journal of Molecular Sciences 23, no. 8: 4422. https://doi.org/10.3390/ijms23084422