
Functional Rescue of Inactivating Mutations of the Human Neurokinin 3 Receptor Using Pharmacological Chaperones

 , , ,
, , ,  , , ,
, , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. NK3R Missense Mutations Impair NKB-Stimulated Inositol Phosphate Generation
2.2. NK3R Missense Mutations Have Variable Effects on Receptor Cell Surface Expression
2.3. Cell Surface Expression of NK3R Mutants Is Rescued by the NK3R Antagonist, M8
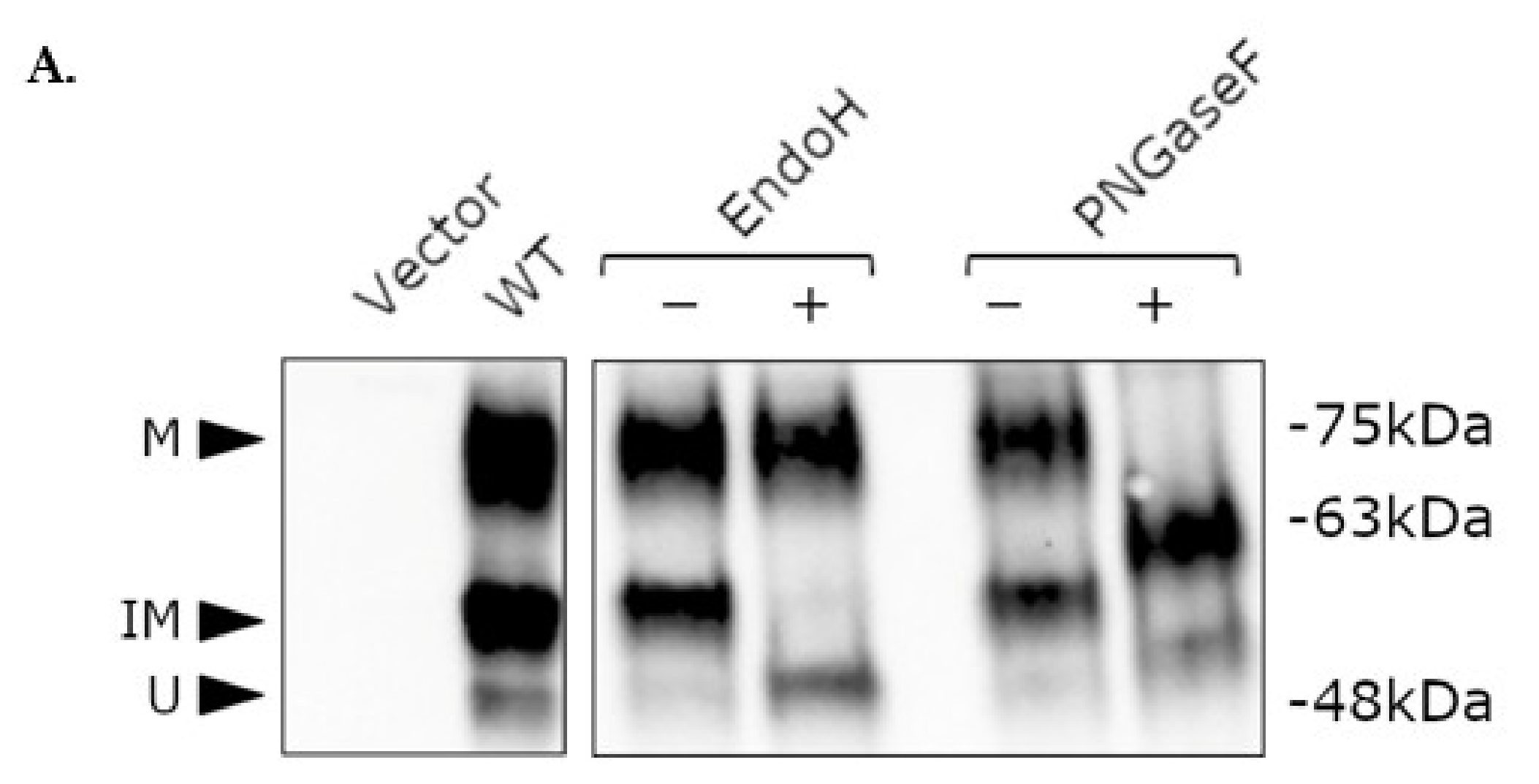
2.4. Post-Translational Modification of WT and Mutant NK3R
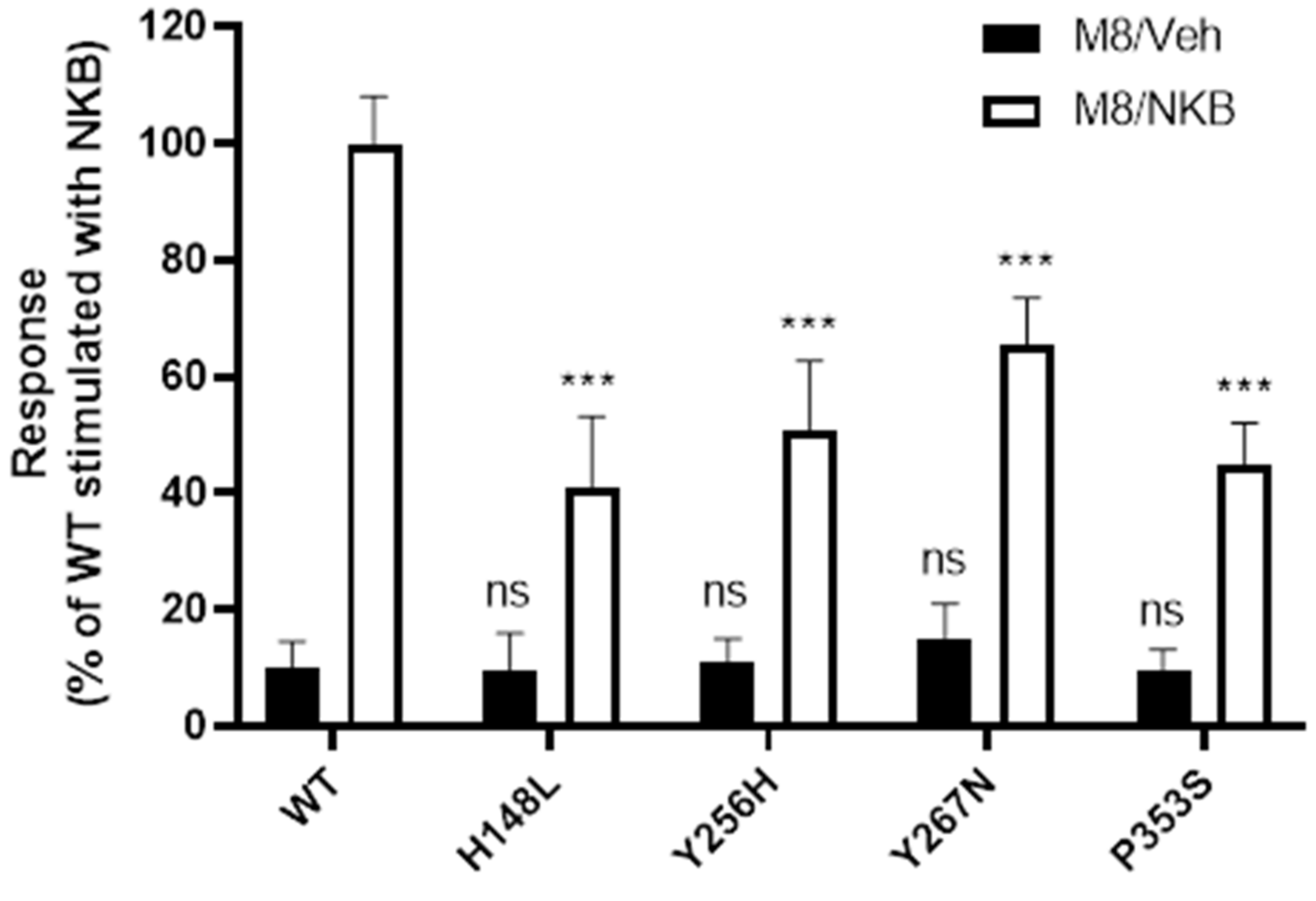
2.5. WT and Mutant NK3R Mutants Rescued by M8 Are Functional
3. Discussion
3.1. NK3R Mutations G93D, H148L, Y256H, Y267N, and P353S Result in Severely Impaired Cell Surface Expression
3.2. M8 Can Restore the Impaired Cell Surface Expression of H148L, Y256H, Y267H, and P353S Mutant NK3Rs
3.3. Increased Cell Surface Expression Restores Function to the Rescued Mutant Receptors
3.4. Y315C and R295S Mutant NK3Rs Have Good to Moderate Cell Surface Expression but Impaired NKB Binding and/or Signalling
4. Materials and Methods
4.1. Materials
4.2. Site-Directed Mutagenesis
4.3. Cell Culture and Transfection
4.4. Inositol Phosphate Accumulation Assay
4.5. Receptor ELISA
4.6. Western Blotting and Deglycosylation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, M.D.; Percy, M.E.; McIntyre Burnham, W.; Cole, D.E. G Protein-Coupled Receptors Disrupted in Human Genetic Disease. Methods Mol. Biol. 2008, 448, 109–137. [Google Scholar] [PubMed]
- Ulloa-Aguirre, A.; Zarinan, T.; Dias, J.A.; Conn, P.M. Mutations in G Protein-Coupled Receptors that Impact Receptor Trafficking and Reproductive Function. Mol. Cell. Endocrinol. 2014, 382, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.X.; Conn, P.M. Chaperoning G Protein-Coupled Receptors: From Cell Biology to Therapeutics. Endocr. Rev. 2014, 35, 602–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulloa-Aguirre, A.; Conn, P.M. Targeting of G Protein-Coupled Receptors to the Plasma Membrane in Health and Disease. Front. Biosci. (Landmark Ed.) 2009, 14, 973–994. [Google Scholar] [CrossRef] [Green Version]
- Young, B.; Wertman, J.; Dupre, D.J. Regulation of GPCR Anterograde Trafficking by Molecular Chaperones and Motifs. Prog. Mol. Biol. Transl. Sci. 2015, 132, 289–305. [Google Scholar]
- Tao, Y.X. Inactivating Mutations of G Protein-Coupled Receptors and Diseases: Structure-Function Insights and Therapeutic Implications. Pharmacol. Ther. 2006, 111, 949–973. [Google Scholar] [CrossRef]
- Cortez, L.; Sim, V. The Therapeutic Potential of Chemical Chaperones in Protein Folding Diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Perlmutter, D.H. Chemical Chaperones: A Pharmacological Strategy for Disorders of Protein Folding and Trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef]
- Wang, Y.J.; Di, X.J.; Mu, T.W. Using Pharmacological Chaperones to Restore Proteostasis. Pharmacol. Res. 2014, 83, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Janovick, J.A.; Maya-Núñez, G.; Ulloa-Aguirre, A.; Huhtaniemi, I.T.; Dias, J.A.; Verbost, P.; Conn, P.M. Increased Plasma Membrane Expression of Human Follicle-Stimulating Hormone Receptor by a Small Molecule Thienopyr(Im)Idine. Mol. Cell. Endocrinol. 2009, 298, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Finch, A.R.; Caunt, C.J.; Armstrong, S.P.; McArdle, C.A. Plasma Membrane Expression of Gonadotropin-Releasing Hormone Receptors: Regulation by Peptide and Nonpeptide Antagonists. Mol. Endocrinol. 2010, 24, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernier, V.; Morello, J.P.; Zarruk, A.; Debrand, N.; Salahpour, A.; Lonergan, M.; Arthus, M.F.; Laperriere, A.; Brouard, R.; Bouvier, M.; et al. Pharmacologic Chaperones as a Potential Treatment for X-Linked Nephrogenic Diabetes Insipidus. J. Am. Soc. Nephrol. 2006, 17, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janovick, J.A.; Stewart, M.D.; Jacob, D.; Martin, L.D.; Deng, J.M.; Stewart, C.A.; Wang, Y.; Cornea, A.; Chavali, L.; Lopez, S.; et al. Restoration of Testis Function in Hypogonadotropic Hypogonadal Mice Harboring a Misfolded GnRHR Mutant by Pharmacoperone Drug Therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 21030–21035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, C.L.; Anderson, R.C.; Kreuchwig, A.; Krause, G.; Katz, A.A.; Millar, R.P. Rescue of Function of Mutant Luteinising Hormone Receptors with Deficiencies in Cell Surface Expression, Hormone Binding and Hormone Signalling. Neuroendocrinology 2020, 111, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Hanyroup, S.; Anderson, R.C.; Nataraja, S.; Yu, H.N.; Millar, R.P.; Newton, C.L. Rescue of Cell Surface Expression and Signalling of Mutant Follicle-Stimulating Hormone Receptors. Endocrinology 2021, 162, bqab134. [Google Scholar] [CrossRef]
- Topaloglu, A.K.; Reimann, F.; Guclu, M.; Yalin, A.S.; Kotan, L.D.; Porter, K.M.; Serin, A.; Mungan, N.O.; Cook, J.R.; Imamoglu, S.; et al. TAC3 and TACR3 Mutations in Familial Hypogonadotropic Hypogonadism Reveal a Key Role for Neurokinin B in the Central Control of Reproduction. Nat. Genet. 2009, 41, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guran, T.; Tolhurst, G.; Bereket, A.; Rocha, N.; Porter, K.; Turan, S.; Gribble, F.M.; Kotan, L.D.; Akcay, T.; Atay, Z.; et al. Hypogonadotropic Hypogonadism due to a Novel Missense Mutation in the First Extracellular Loop of the Neurokinin B Receptor. J. Clin. Endocrinol. Metab. 2009, 94, 3633–3639. [Google Scholar] [CrossRef] [Green Version]
- Gianetti, E.; Tusset, C.; Noel, S.D.; Au, M.G.; Dwyer, A.A.; Hughes, V.A.; Abreu, A.P.; Carroll, J.; Trarbach, E.; Silveira, L.F.; et al. TAC3/TACR3 Mutations Reveal Preferential Activation of Gonadotropin-Releasing Hormone Release by Neurokinin B in Neonatal Life Followed by Reversal in Adulthood. J. Clin. Endocrinol. Metab. 2010, 95, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Francou, B.; Bouligand, J.; Voican, A.; Amazit, L.; Trabado, S.; Fagart, J.; Meduri, G.; Brailly-Tabard, S.; Chanson, P.; Lecomte, P.; et al. Normosmic Congenital Hypogonadotropic Hypogonadism due to TAC3/TACR3 Mutations: Characterization of Neuroendocrine Phenotypes and Novel Mutations. PLoS ONE 2011, 6, e25614. [Google Scholar] [CrossRef] [Green Version]
- Tusset, C.; Noel, S.D.; Trarbach, E.B.; Silveira, L.F.; Jorge, A.A.; Brito, V.N.; Cukier, P.; Seminara, S.B.; Mendonca, B.B.; Kaiser, U.B.; et al. Mutational Analysis of TAC3 and TACR3 Genes in Patients with Idiopathic Central Pubertal Disorders. Arq. Bras. Endocrinol. Metabol. 2012, 56, 646–652. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K.; Mizuno, H.; Tanaka, T.; Togawa, T.; Negishi, Y.; Ohashi, K.; Hori, I.; Izawa, M.; Hamajima, T.; Saitoh, S. Molecular Genetic and Clinical Delineation of 22 Patients with Congenital Hypogonadotropic Hypogonadism. J. Pediatr. Endocrinol. Metab. 2017, 30, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Ruka, K.A.; Burger, L.L.; Moenter, S.M. Regulation of Arcuate Neurons Coexpressing Kisspeptin, Neurokinin B, and Dynorphin by Modulators of Neurokinin 3 and Kappa-Opioid Receptors in Adult Male Mice. Endocrinology 2013, 154, 2761–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, S.; Seminara, S.B.; Plant, T.M. Evidence from the Agonadal Juvenile Male Rhesus Monkey (Macaca Mulatta) for the View that the Action of Neurokinin B to Trigger Gonadotropin-Releasing Hormone Release is Upstream from the Kisspeptin Receptor. Neuroendocrinology 2011, 94, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.; George, J.T.; Tello, J.A.; Francou, B.; Bouligand, J.; Guiochon-Mantel, A.; Brailly-Tabard, S.; Anderson, R.A.; Millar, R.P. Kisspeptin Restores Pulsatile LH Secretion in Patients with Neurokinin B Signaling Deficiencies: Physiological, Pathophysiological and Therapeutic Implications. Neuroendocrinology 2013, 97, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.C.; Letts, P.A.; Krajewski, S.J.; Rance, N.E. Coexpression of Dynorphin and Neurokinin B Immunoreactivity in the Rat Hypothalamus: Morphologic Evidence of Interrelated Function within the Arcuate Nucleus. J. Comp. Neurol. 2006, 498, 712–726. [Google Scholar] [CrossRef]
- Jayasena, C.N.; Comninos, A.N.; Stefanopoulou, E.; Buckley, A.; Narayanaswamy, S.; Izzi-Engbeaya, C.; Abbara, A.; Ratnasabapathy, R.; Mogford, J.; Ng, N.; et al. Neurokinin B Administration Induces Hot Flushes in Women. Sci. Rep. 2015, 5, 8466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.W.; Wang, Y.; Chu, Y.X. Tacr3/NK3R: Beyond their Roles in Reproduction. ACS Chem. Neurosci. 2020, 11, 2935–2943. [Google Scholar] [CrossRef]
- Millar, R.P.; Newton, C.L. Current and Future Applications of GnRH, Kisspeptin and Neurokinin B Analogues. Nat. Rev. Endocrinol. 2013, 9, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.M.; Carling, R.W.; Chicchi, G.G.; Crawforth, J.; Hutson, P.H.; Jones, A.B.; Kelly, S.; Marwood, R.; Meneses-Lorente, G.; Mezzogori, E.; et al. N’,2-Diphenylquinoline-4-Carbohydrazide Based NK3 Receptor Antagonists II. Bioorganic Med. Chem. Lett. 2006, 16, 5752–5756. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pandy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keseru, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR Sequence, Structure and Function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef]
- Leidenheimer, N.J.; Ryder, K.G. Pharmacological Chaperoning: A Primer on Mechanism and Pharmacology. Pharmacol. Res. 2014, 83, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robben, J.H.; Sze, M.; Knoers, N.V.; Deen, P.M. Functional Rescue of Vasopressin V2 Receptor Mutants in MDCK Cells by Pharmacochaperones: Relevance to Therapy of Nephrogenic Diabetes Insipidus. Am. J. Physiol. Renal Physiol. 2007, 292, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janovick, J.A.; Goulet, M.; Bush, E.; Greer, J.; Wettlaufer, D.G.; Conn, P.M. Structure-Activity Relations of Successful Pharmacologic Chaperones for Rescue of Naturally Occurring and Manufactured Mutants of the Gonadotropin-Releasing Hormone Receptor. J. Pharmacol. Exp. Ther. 2003, 305, 608–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, V.M. Interactions between Kisspeptins and Neurokinin B. Adv. Exp. Med. Biol. 2013, 784, 325–347. [Google Scholar] [PubMed] [Green Version]
- Ulloa-Aguirre, A.; Michael Conn, P. Pharmacoperones: A New Therapeutic Approach for Diseases Caused by Misfolded G Protein-Coupled Receptors. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Beerepoot, P.; Nazari, R.; Salahpour, A. Pharmacological Chaperone Approaches for Rescuing GPCR Mutants: Current State, Challenges, and Screening Strategies. Pharmacol. Res. 2017, 117, 242–251. [Google Scholar] [CrossRef]
- Prague, J.K. Neurokinin 3 Receptor Antagonists—Prime Time? Climacteric 2021, 24, 25–31. [Google Scholar] [CrossRef]
- Young, J.; Bouligand, J.; Francou, B.; Raffin-Sanson, M.L.; Gaillez, S.; Jeanpierre, M.; Grynberg, M.; Kamenicky, P.; Chanson, P.; Brailly-Tabard, S.; et al. TAC3 and TACR3 Defects Cause Hypothalamic Congenital Hypogonadotropic Hypogonadism in Humans. J. Clin. Endocrinol. Metab. 2010, 95, 2287–2295. [Google Scholar] [CrossRef] [Green Version]
- Morello, J.P.; Salahpour, A.; Laperrière, A.; Bernier, V.; Arthus, M.F.; Lonergan, M.; Petäjä-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological Chaperones Rescue Cell-Surface Expression and Function of Misfolded V2 Vasopressin Receptor Mutants. J. Clin. Investig. 2000, 105, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Newton, C.L.; Anderson, R.C.; Katz, A.A.; Millar, R.P. Loss-of-Function Mutations in the Human Luteinizing Hormone Receptor Predominantly Cause Intracellular Retention. Endocrinology 2016, 157, 4364–4377. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Analysis and Refinement of Criteria for Predicting the Structure and Relative Orientations of Transmembranal Helical Domains. Biophys. J. 1992, 62, 107–109. [Google Scholar] [CrossRef] [Green Version]
- Malherbe, P.; Bissantz, C.; Marcuz, A.; Kratzeisen, C.; Zenner, M.T.; Wettstein, J.G.; Ratni, H.; Riemer, C.; Spooren, W. Me-Talnetant and Osanetant Interact within Overlapping but Not Identical Binding Pockets in the Human Tachykinin Neurokinin 3 Receptor Transmembrane Domains. Mol. Pharmacol. 2008, 73, 1736–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, S.D.; Abreu, A.P.; Xu, S.; Muyide, T.; Gianetti, E.; Tusset, C.; Carroll, J.; Latronico, A.C.; Seminara, S.B.; Carroll, R.S.; et al. TACR3 Mutations Disrupt NK3R Function through Distinct Mechanisms in GnRH-Deficient Patients. FASEB J. 2014, 28, 1924–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tansky, M.F.; Pothoulakis, C.; Leeman, S.E. Functional Consequences of Alteration of N-Linked Glycosylation Sites on the Neurokinin 1 Receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 10691–10696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goth, C.K.; Petaja-Repo, U.E.; Rosenkilde, M.M. G Protein-Coupled Receptors in the Sweet Spot: Glycosylation and Other Post-Translational Modifications. ACS Pharmacol. Transl. Sci. 2020, 3, 237–245. [Google Scholar] [CrossRef]
- Petaja-Repo, U.E.; Hogue, M.; Laperriere, A.; Walker, P.; Bouvier, M. Export from the Endoplasmic Reticulum Represents the Limiting Step in the Maturation and Cell Surface Expression of the Human Delta Opioid Receptor. J. Biol. Chem. 2000, 275, 13727–13736. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Filipeanu, C.M.; Duvernay, M.T.; Wu, G. Regulation of G Protein-Coupled Receptor Export Trafficking. Biochim. Biophys. Acta Biomembr. 2007, 1768, 853–870. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, M.; Hawtin, S.R. Glycosylation of G-Protein-Coupled Receptors for Hormones Central to Normal Reproductive Functioning: Its Occurrence and Role. Hum. Reprod. Update 1999, 5, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Ganjiwale, A.D.; Rao, G.S.; Cowsik, S.M. Molecular Modeling of Neurokinin B and Tachykinin NK(3) Receptor Complex. J. Chem. Inf. Model. 2011, 51, 2932–2938. [Google Scholar] [CrossRef]
- Neumann, S.; Krause, G.; Claus, M.; Paschke, R. Structural Determinants for G Protein Activation and Selectivity in the Second Intracellular Loop of the Thyrotropin Receptor. Endocrinology 2005, 146, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Gether, U. Uncovering Molecular Mechanisms Involved in Activation of G Protein-Coupled Receptors. Endocr. Rev. 2000, 21, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Diversity and Modularity of G Protein-Coupled Receptor Structures. Trends Pharmacol. Sci. 2012, 33, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Gilda, J.E.; Gomes, A.V. The Necessity of and Strategies for Improving Confidence in the Accuracy of Western Blots. Expert Rev. Proteom. 2014, 11, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, R.C.; Hanyroup, S.; Song, Y.B.; Mohamed-Moosa, Z.; van den Bout, I.; Schwulst, A.C.; Kaiser, U.B.; Millar, R.P.; Newton, C.L. Functional Rescue of Inactivating Mutations of the Human Neurokinin 3 Receptor Using Pharmacological Chaperones. Int. J. Mol. Sci. 2022, 23, 4587. https://doi.org/10.3390/ijms23094587
Anderson RC, Hanyroup S, Song YB, Mohamed-Moosa Z, van den Bout I, Schwulst AC, Kaiser UB, Millar RP, Newton CL. Functional Rescue of Inactivating Mutations of the Human Neurokinin 3 Receptor Using Pharmacological Chaperones. International Journal of Molecular Sciences. 2022; 23(9):4587. https://doi.org/10.3390/ijms23094587
Chicago/Turabian StyleAnderson, Ross C., Sharika Hanyroup, Yong Bhum Song, Zulfiah Mohamed-Moosa, Iman van den Bout, Alexis C. Schwulst, Ursula B. Kaiser, Robert P. Millar, and Claire L. Newton. 2022. "Functional Rescue of Inactivating Mutations of the Human Neurokinin 3 Receptor Using Pharmacological Chaperones" International Journal of Molecular Sciences 23, no. 9: 4587. https://doi.org/10.3390/ijms23094587
APA StyleAnderson, R. C., Hanyroup, S., Song, Y. B., Mohamed-Moosa, Z., van den Bout, I., Schwulst, A. C., Kaiser, U. B., Millar, R. P., & Newton, C. L. (2022). Functional Rescue of Inactivating Mutations of the Human Neurokinin 3 Receptor Using Pharmacological Chaperones. International Journal of Molecular Sciences, 23(9), 4587. https://doi.org/10.3390/ijms23094587