1. Introduction
Glycogen storage disease type V (GSDV) (OMIM#232600), also known as McArdle disease, is a rare autosomal recessive myopathy caused by biallelic pathogenic mutations in the
PYGM gene [
1] that result in deficiency of the skeletal muscle isoform of glycogen phosphorylase (or ‘myophosphorylase’, PYGM) [
2]. GSDV has been reported to have an estimated prevalence of 1 in 100,000—350,000 people [
2,
3,
4,
5].
Because PYGM catalyzes the first rate-limiting step of glycogen metabolism (i.e., removal of terminal alpha-1,4-glycosidic bonds from the outer branches of this molecule to release glucose-1-phosphate), deficiency of this enzyme leads to a block in the use of glycogen as an energy source for muscle contraction [
6]. Typical clinical features consist of muscle ‘crises’ of pain and fatigue, together with tachycardia during the first minutes of dynamic exercise (e.g., brisk walking) that are attenuated after 7–10 min have elapsed—the so-called ‘second wind’ phenomenon [
2,
7]. These episodes of early exercise intolerance are frequently accompanied by severe muscle contractures, potentially leading to rhabdomyolysis and subsequent myoglobinuria, as reflected by ‘dark urines’. Yet, another feature of the disease is a persistent status of muscle damage—(as reflected by very high circulating levels of intra-muscle proteins such as creatine kinase [CK]), even in the absence of physical exercise on the previous day(s) [
8].
More than 170 pathogenic mutations (including missense, nonsense, in-frame, frameshift, and splicing variants) have been identified in the
PYGM gene that cause McArdle disease [
9,
10]. Most of these mutations result in a total absence of PYGM activity [
11] in the patients’ muscle tissue, except for two patients carrying deep-intronic mutations in compound heterozygosity that led to some residual (~1% of normal) enzyme activity, with subsequent amelioration in clinical phenotype [
12]. There is no association between the
PYGM genotype and disease phenotype, since patients with the same mutation(s) can show quite different degrees of clinical severity [
13]. The pathobiology of GSDV is not fully understood, but it seems that the potential molecular consequences of the lack of glycogenolytic–derived ATP involve not only the expected energetic deficit for actin-myosin cross bridging, but also impairments in membrane pump function, excitation–contraction coupling, and sarcolemmal excitability [
14].
In an attempt to identify potential muscle protein biomarkers and gain insight into the pathobiology of GSDV, we analyzed the targeted proteome in skeletal muscle biopsies obtained from both patients with histochemical and genetic diagnoses of GSDV and healthy controls. In this regard, since skeletal muscle is the only tissue that is clinically affected in all patients with GSDV, the control tissue was skeletal muscle biopsies from aged and sex-matched healthy individuals with normal PYGM activity and no signs of the typical features of McArdle disease, such as frequent exercise-induced contractures or persistent muscle damage in the absence of prior exertion.
We first used isobaric tags for relative and absolute quantitation (iTRAQ) analysis [
15] to compare the muscle protein expression in patients vs. controls. This was followed by a systems biology network-based approach to identify key proteins involved in distinct pathways that could be related to the GSDV phenotype, such as the breakdown of muscle fibers, muscle contractures, and impairment in calcium homeostasis or in other physiological processes of the skeletal muscle. To this end, we applied the therapeutic performance mapping system (TPMS) machine learning-based technology, particularly by applying artificial neuronal networks (ANNs) that were ‘trained’ using the human protein network and drug-pathophysiology knowledge [
16,
17]. This technology has proven useful to identify non-obvious functional relationships for drug repurposing purposes [
18,
19,
20] and biological data analysis and prioritization of proteins according to documented relationships with pathophysiological processes [
21,
22,
23], especially in rare diseases or when sample sizes are limited. After the prioritization process, levels of the selected candidate proteins were analyzed using Western blot analyses.
Our results indicate that in addition to PYGM, myosin 1 (MYH-1), tropomyosin alpha-1 chain (TPM1), sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (ATP2A1, also abbreviated as SERCA1), troponin isoforms (troponin I2, fast skeletal type [TNNI2] and troponin T3, fast skeletal type [TNNT3]), and alpha-actinin-3 (ACTN3) show a relationship with GSDV, with their levels reduced in the skeletal muscle tissue of GSDV patients with respect to healthy controls. Most of these proteins are involved in muscle contractures associated with altered calcium homeostasis.
2. Results
The main characteristics of the patients are shown in
Table 1 and
Table 2. Sex distribution (50% and 62.5% female in patients and controls; Chi-square test’s
p = 0.625) and mean (± SD) age (patients: 38 ± 12 years; controls: 40 ± 9 years; Mann–Whitney’s U
p = 0.711) did not differ between the two groups.
By quantitative proteome analysis of skeletal muscle biopsies obtained from the
Biceps brachii or
Vastus lateralis of eight GSDV patients and eight healthy controls using iTRAQ labeling followed by reversed-phase liquid chromatography-mass spectrometry (RP-LC-MS/MS), 178 proteins were identified. The patient and healthy control samples were separately pooled, and parallel double labeling was performed for each pool, resulting in two label values per group (113 and 115 for patients and 114 and 116 for controls); all values were referenced to the values of the 113 patients’ pool (
Supplementary Table S1).
The peptide value distribution for each protein with peptide number >10 was compared between control and patient pool values, respectively, to obtain a total of 21 proteins with comparable control pool values, on the one hand, and differences between controls and patients, on the other (
Table 3). These results were used to set a control/patient value ratio-based threshold, considering the mean of this value for these 21 proteins (= 1.675).
Next, we detected the most differentially expressed proteins by calculating the control/patient value ratio of the global data (i.e., for all 178 proteins detected [
Supplementary Table S1] regardless of the number of peptides measured) and identified 15 proteins with control/patient values ratio >1.676 (
Table 4).
To allow analyses with TPMS technology, all data were mapped to 14 unique reviewed SwissProtKB entries (
Table 4). Nine of the fourteen proteins exhibited at least a two-fold higher change in one of the control pools compared to the 113-labeled patient pool, which was used as a reference for labeling the rest of the proteins (indicated as bold values in
Table 4).
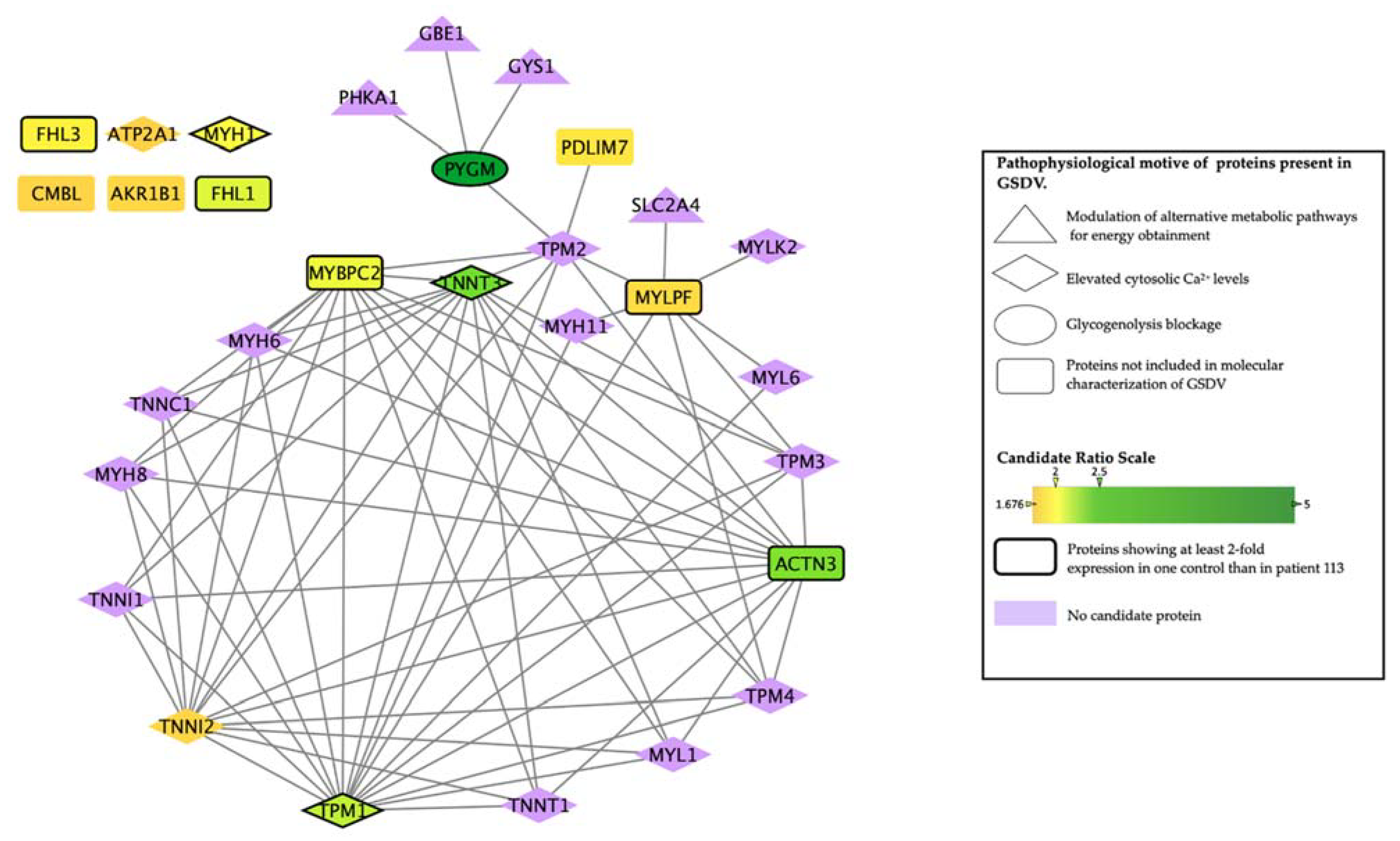
The possible relationship between the most differentially expressed proteins (
Table 4) and GSDV attending to their ‘molecular characterization’ was evaluated by means of ANNs (
Table 5 and
Supplementary Table S3 show results considering GSDV as a whole or considering the different motives separately, respectively). Attending to the associated
p-values, we sorted the ANN ranking score into four categories: ‘very strong’ (
p < 0.01), ‘strong’ (
p < 0.05), ‘medium-strong’ (
p < 0.25), and ‘weak’ (
p > 0.25) (
Supplementary Table S4). Three proteins, ATP2A1, MYH1, and TPM1, showed a very strong relationship with GSDV (
Table 5). These three proteins were part of the functional motif
elevated cytosolic calcium levels, and specifically of the
persistent contraction of muscle cell sub-motif for MYH1 and TPM1 (
Supplementary Tables S2 and S3). The troponin isoforms, TNNI2 and TNNT3, displayed a strong relationship with GSDV (
Table 5) and were also effectors of the sub-motif
persistent contraction of muscle cells. All the proteins were related to muscle structure and activity and showed a stronger relationship with GSDV definition than the enzyme PYGM, the defective protein in GSDV, which presented a medium-strong score with GSDV molecular characterization and was assigned as an effector of the glycogenolytic pathway.
The PDZ and LIM domain protein 7 (PDLIM7) and alpha-actinin-3 (ACTN3) showed a medium-strong score, and the rest of the evaluated proteins showed a low probability of being related to GSDV in a molecular-dependent manner (
Table 5), according to the used molecular characterization.
After evaluating the relationship between the most differentially expressed proteins and each GSDV motif, as described by molecular characterization (
Supplementary Table S3), we observed that the motif that exhibited the highest probability of a relationship with the available data was
elevated cytosolic calcium levels, and particularly the submotif
persistent contraction of muscle cell. Most of these proteins showed a strong or medium-strong probability of a relationship with the motif and submotif. In fact, for all the candidate proteins, the highest probability score was observed for
elevated cytosolic calcium levels.
To further understand the intermolecular relationships identified by ANNs, we generated a protein interactome with the human protein network used for model construction and based on publicly available sources. This allowed us to identify the interaction between the most differentially expressed (‘candidate’) proteins and the effector proteins identified as important in GSDV molecular characterization (
Table 5,
Figure 1). Most of the candidates showed an interaction with effectors of the biological motives
elevated cytosolic calcium levels-persistent contraction of muscle cell (
Supplementary Tables S3 and S5). However, two of the most differentially expressed proteins, the skeletal muscle isoform of the myosin regulatory light chain 2 (MYLPF) and PYGM, interacted with proteins belonging to the
modulation of alternative metabolic pathways for energy obtainment-increased glucose uptake motif. In addition, according to the databases used (see topological analysis in the methods section), six of the most differentially expressed proteins (i.e., MYH1, ATP2A1, isoform 1 of four and a half LIM domains protein 1 (FHL1), four and a half LIM domains protein 3 (FHL3), aldose reductase (AKR1B1) and carboxymethylenebutenolidase homolog (CMBL)) did not directly interact with any of the GSDV effectors nor with any other most differentially expressed proteins (
Table 5).
From the list of the most differentially expressed proteins, myosin light chain, phosphorylatable, fast skeletal muscle (MYLPF, Q96A32), and myosin binding protein (MYBPC2, Q14324) were not predicted to be related to GSDV (weak relationship in
Table 5); however, they appeared highly connected to proteins within the
elevated cytosolic Ca2+ levels motif (
Figure 1), which could explain the medium-strong signal detected between these proteins and this motif (
Supplementary Table S3) despite not being its effectors.
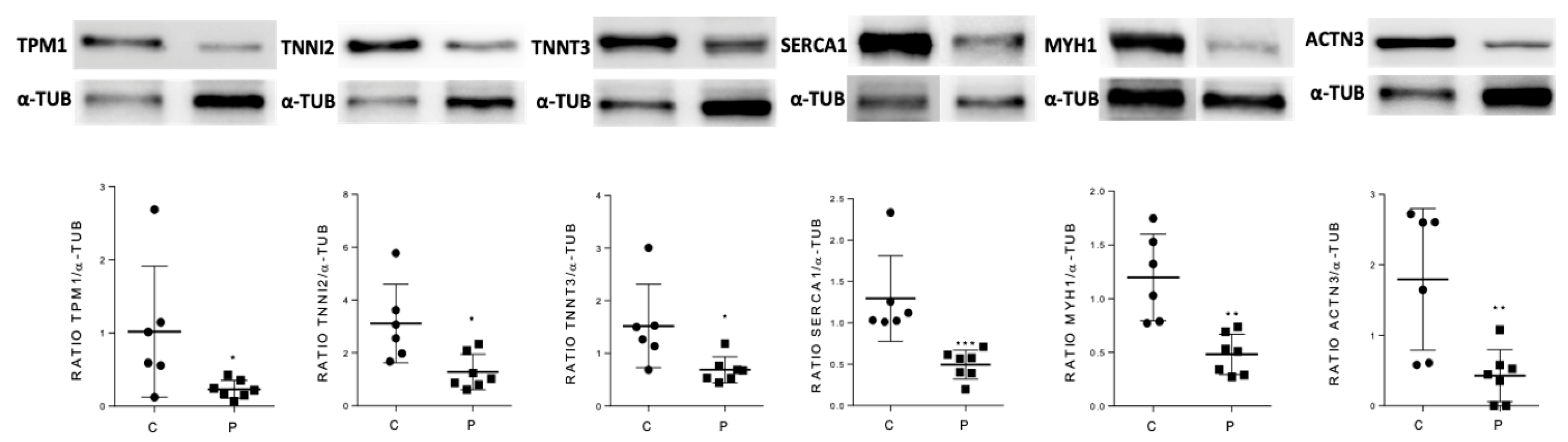
To validate the predictive results obtained using the ANN analysis strategy, skeletal muscle levels of a selected group of candidate proteins were also analyzed by Western blot in GSDV patients and healthy controls. We selected as candidates the most differentially expressed proteins that were classified in the ‘very strong’ and ‘strong’ categories according to their relationship with GSDV as a whole (
Table 5): MYH1, ATP2A1, TPM1, TNNI2, and TNNT3. Besides these proteins, ACTN3 was also considered a candidate and analyzed despite being ranked in the medium-strong category, due to two relevant reasons: (i) it has been documented to interact with PYGM and implicated in altered muscle calcium handling in the
Actn3 deficient (knockout) mouse model [
27], and (ii) at least in female patients,
ACTN3 genotypes might contribute to explaining individual variability in the phenotypic manifestation of this disorder [
28,
29]. We showed that the expression levels of all tested candidates (MYH1, ATP2A1, TPM1, TNNI2, TNNT3, and ACTN3) were significantly lower in patients than in controls (
Figure 2 and
Supplementary Figure S1).
3. Discussion
GSDV is a metabolic myopathy typically characterized by exercise intolerance (i.e., muscle pain and early exertional fatigue). If the exercise stress is not reduced or halted, severe muscle contractures (beyond the usual baseline state of ‘persistent’ muscle contraction and damage) and eventual rhabdomyolysis might occur, which in some cases, could result in acute renal failure [
4,
30]. Although the knowledge of the molecular and pathophysiologic mechanisms of GSDV has improved during the last two decades, particularly with insights provided by clinical, molecular, or physiological studies in patients [
4,
14,
24,
31,
32,
33,
34,
35], as well as by studies in preclinical models [
21,
34,
36,
37,
38,
39,
40], there is still no explanation (at least at the molecular level) for some recognized clinical features of the disease, notably the persistent muscle damage in the absence of previous physical exercise [
8].
We therefore aimed at investigating in depth the muscle proteome and the molecular networks associated with muscle dysfunction in GSDV patients in an attempt to identify key muscle proteins as biomarkers that could help to understand the underlying molecular mechanisms of muscle dysfunction or damage. To the best of our knowledge, this question has not been explored previously. In a case-control design with muscle biopsies from histochemical and genetically proven GSDV patients and from healthy controls, we assessed quantitative protein expression using the iTRAQ technique and then performed a systems biology-based strategy, particularly applying ANNs and topology interactome networks to identify the best candidates. Our analysis suggested that some of the identified candidate proteins are related to GSDV disease predominantly through the motif persistent contraction of muscle cells due to elevated cytosolic calcium levels, with the proteins ACTN3, ATP2A1, MYH1, TNNT3, TPM1, and TNNI2 showing the highest predictive values among all the proteins evaluated. Furthermore, the topological analysis indicated that the candidate proteins identified in this study interact with proteins involved in the persistent contraction of muscle cells due to elevated cytosolic calcium levels and the modulation of alternative metabolic pathways for energy obtainment.
The levels of ACTN3, ATP2A1, MYH1, TNNT3, TPM1, and TNNI2 proteins were significantly lower in the skeletal muscle of patients compared with healthy controls. MYH1 is a skeletal muscle protein that, in coordination with actin, plays an essential role in the generation of energy for muscle contraction through ATP hydrolysis [
41]. ATP2A1, the sarcoplasmic/endoplasmic reticulum calcium ATPase 1 (previously known as SERCA1), is a membrane protein that is responsible for the transport of calcium from the sarcoplasm back into the sarcoplasmic reticulum after each sarcomeric contraction, and whose function is dependent on the energy delivered by ATP hydrolysis. Likewise, ATPA21 contributes to the excitation/contraction balance involved in muscle activity [
42]. A decrease in ATP2A1 levels would result in an impairment in the reuptake of calcium back into the sarcoplasmic reticulum after each contraction, with subsequent accumulation of this ion in the sarcoplasm and impairment of muscle fiber relaxation—that is, permanent muscle contraction and muscle contractures. Interestingly, besides the association of primary pathogenic genetic variants in the
ATP2A1 gene with Brody myopathy (OMIM#601003, a rare autosomal recessive disorder characterized by painless muscle cramping and exercise-induced impaired muscle relaxation) [
43], other conditions linked with aging, neurodegeneration, and muscular dystrophy also depress ATP2A1 function with the potential to impair intracellular calcium homeostasis and contribute to muscle atrophy and weakness [
42]. There is some controversy on how to assess calcium homeostasis in different human diseases since most research has been performed in murine models [
44,
45,
46,
47]. On the other hand, the stability of actin filaments in the muscle fibers is ensured by the function of tropomyosin (TPM1), which, in association with the troponin complex (TNNI2 and TNNT3), plays a key role in the regulation of calcium-dependent interactions during muscle contraction [
48]. In addition, ACTN3 plays an important role in the stability of the contractile apparatus at the Z-line, where this protein cross-links and anchors actin filaments [
49]. Therefore, our findings suggest that decreased expression of the aforementioned proteins in GSDV could be associated, at least in part, with the altered muscle contractile function and a probable alteration of muscle calcium kinetics in this disorder. On the other hand, PYGM could also be involved not only in energy generation from glycogen breakdown, but also in the O-linked β-N-acetylglucosamine (O-GlcNa)c post-translational modifications of some proteins [
6,
50]. In this effect, O-GlcNAcylation plays an important role in several skeletal muscle functions, including optimal modulation of calcium homeostasis in fibers [
51,
52].
Our study is limited by the small sample size, although we believe this is justifiable in the context of a rare condition such as McArdle disease. We also failed to collect all the samples from the same muscle, although the vast majority of samples corresponded to the
Biceps brachii, and the proportion of muscle type (i.e., 6/2 for
Biceps brachii/
Vastus lateralis) was identical in patients and healthy controls. Importantly, our approach also lacked a comparison group of patients with similar features to those of McArdle disease, such as muscle contractures—although we are not aware of any neuromuscular condition where muscle contractures are as frequent or persistent as in McArdle disease—and therefore we cannot address if the detected differentially expressed proteins are primarily or secondarily regulated. In addition, it must be kept in mind that with regard to potential biomarkers of McArdle disease, our findings must be viewed as mechanistic—hopefully providing useful insights and framework for future research—rather than practical ones since muscle biopsies represent an invasive procedure and the molecular techniques used here are not easily available in any center. In turn, the method of RP-LC-MS/MS used here (or LC-MS/MS in general) is currently the most effective tool to discover and quantify the human proteome and represents an essential approach for the study of biological systems that is, in fact, routinely applied for diverse applications beyond relative or absolute proteome quantification, including biomarker discovery [
53].
In conclusion, while keeping in mind the aforementioned limitations, our findings suggest some candidate proteins as potential biomarkers of GSDV. Our results provide a framework for future studies aimed at elucidating the molecular mechanisms by which PYGM controls the expression of the most relevant identified proteins.


 ,
, 








{kind=link}
{kind=link}