
Notch–Sox9 Axis Mediates Hepatocyte Dedifferentiation in KrasG12V-Induced Zebrafish Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Induction of Hepatocellular Carcinoma by DOX-Activated Tet-On System in Zebrafish
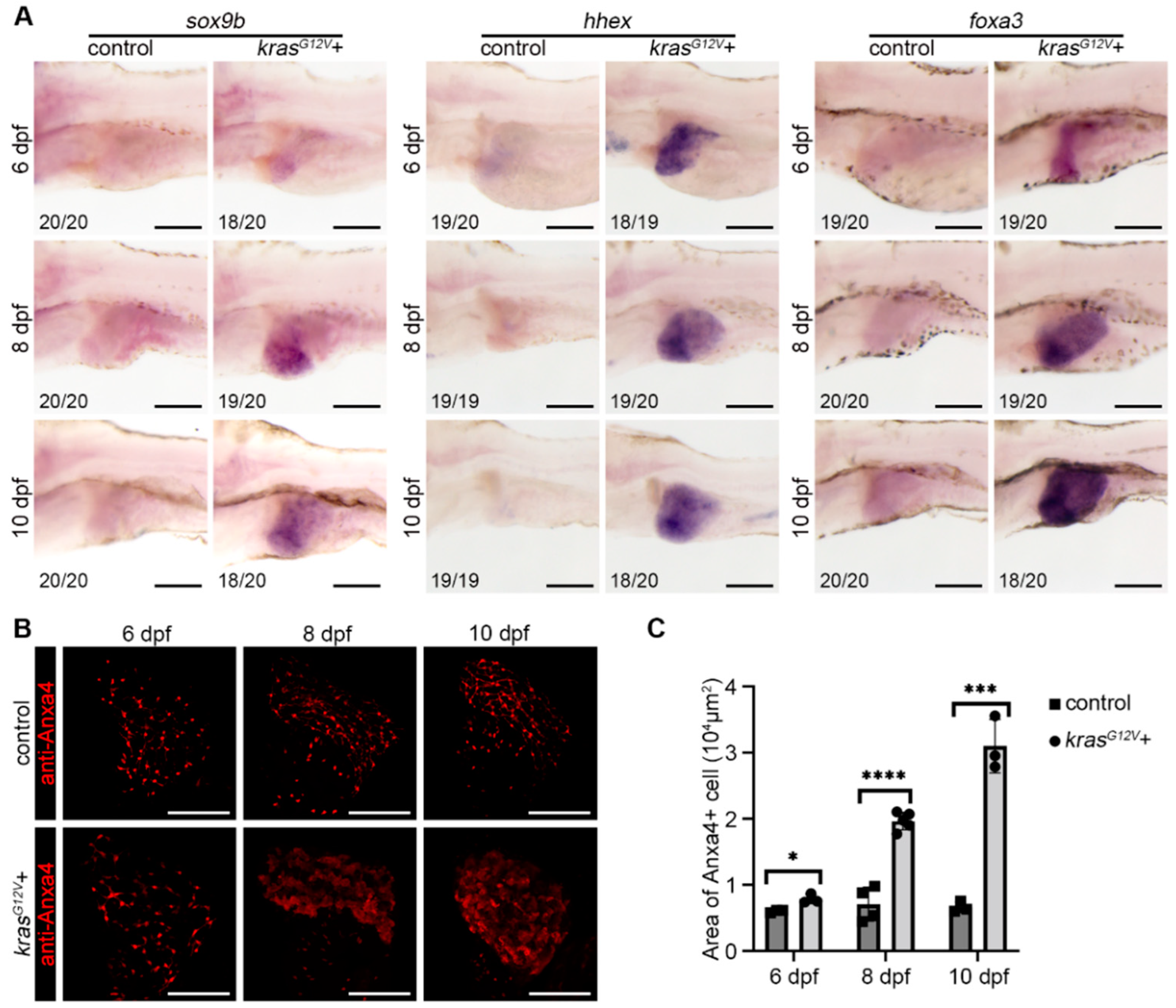
2.2. Hepatocyte Dedifferentiation Occurred after Hepatocarcinogenesis
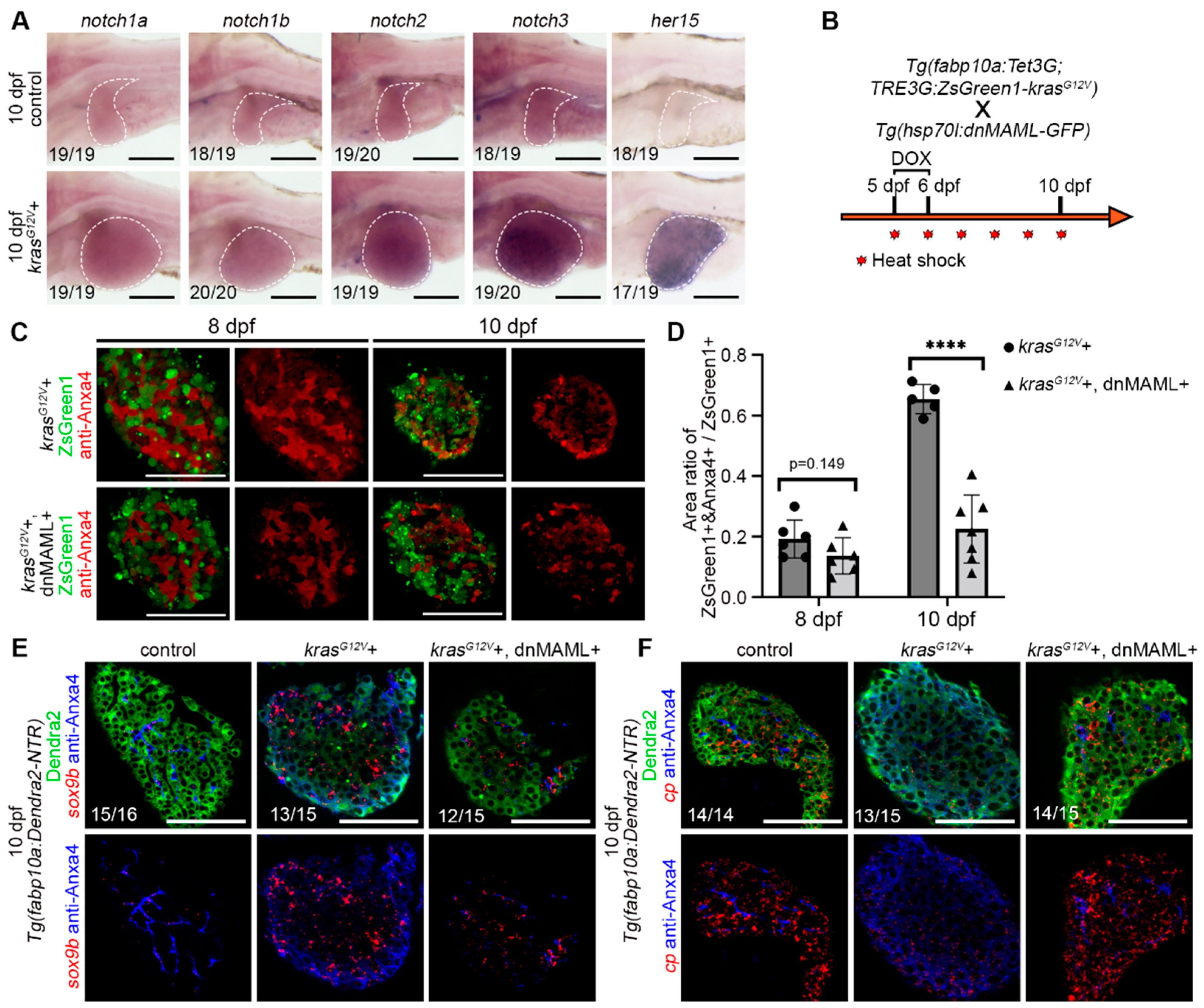
2.3. Hepatocyte Dedifferentiation was Suppressed by Inhibition of Notch Signaling after Hepatocarcinogenesis
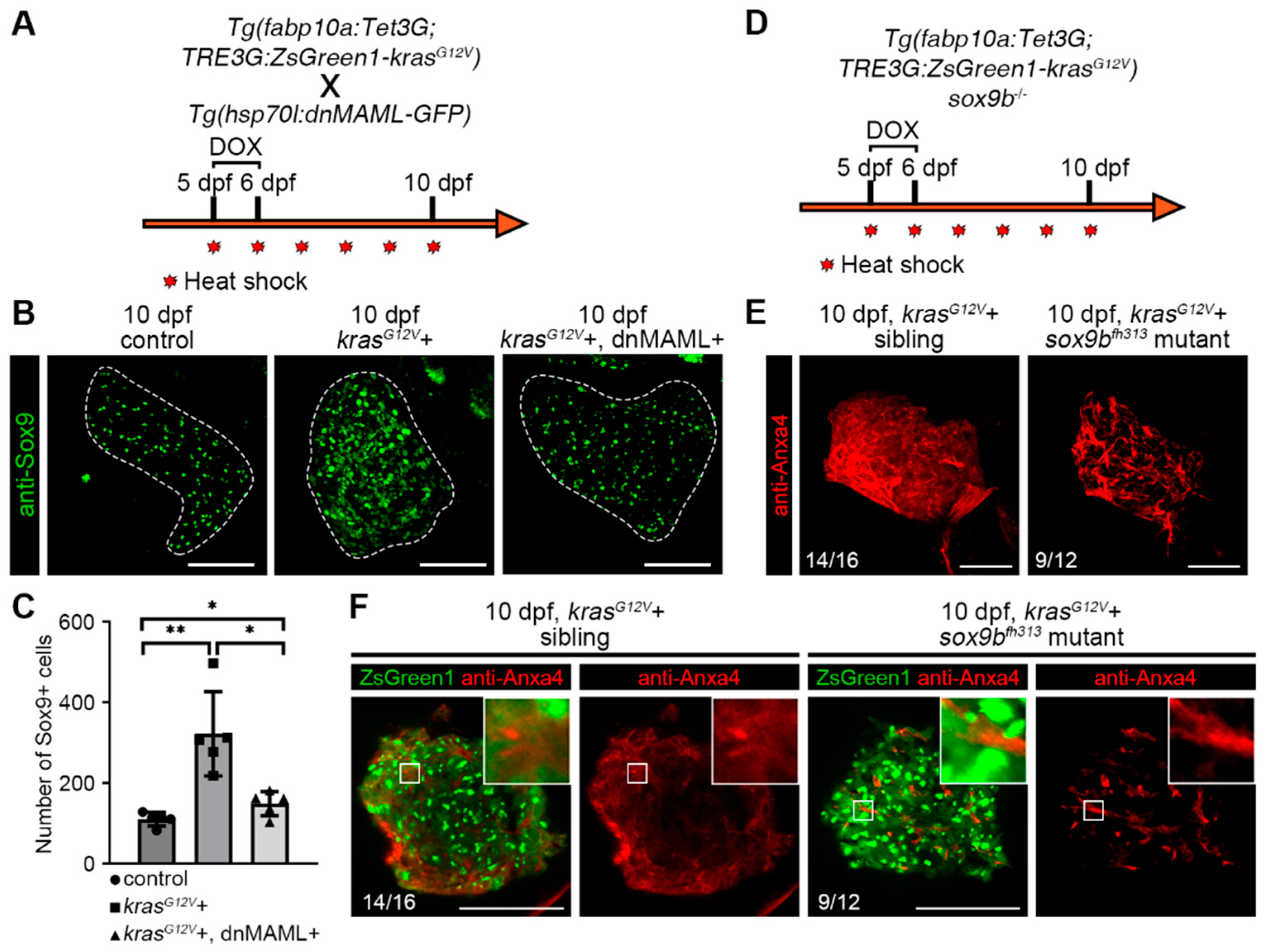
2.4. Notch–Sox9 Axis Mediates Hepatocyte Dedifferentiation
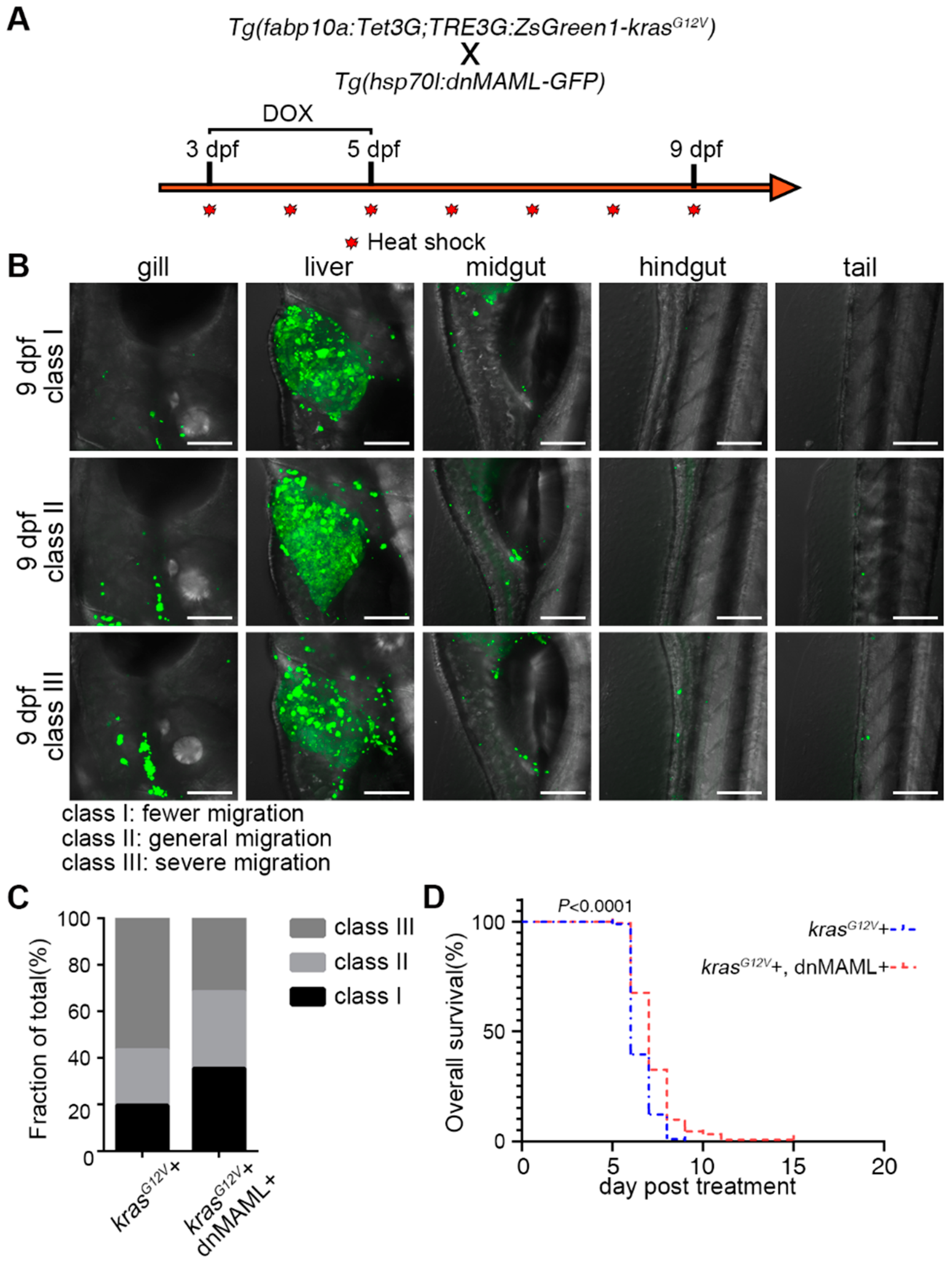
2.5. Inhibition of Notch Signaling Ameliorated Symptoms of Liver Cancer
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Zebrafish Lineage
4.3. Whole-Mount In Situ Hybridization
4.4. Fluorescent In Situ Hybridization Coupled with Antibody Staining Assays
4.5. Antibody Staining
4.6. Generation of Transgenic Lines for Induction Experiments
4.7. DOX Treatment and Heat Shock
4.8. Paraffin Sectioning and Staining
4.9. Percent Fibrosis Statistics
4.10. Data Collection and Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Chen, K.F.; Chen, P.J. Treatment of Liver Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a021535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wong, P.P.; Sjeklocha, L.; Steer, C.J.; Sahin, M.B. Mature hepatocytes exhibit unexpected plasticity by direct dedifferentiation into liver progenitor cells in culture. Hepatology 2012, 55, 563–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell. 2014, 15, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, G.P.; Pinelli, D.; Di Benedetto, F.; Marini, E.; Corno, V.; Guizzetti, M.; Aluffi, A.; Zambelli, M.; Fagiuoli, S.; Luca, M.G.; et al. Predictive value of nodule size and differentiation in HCC recurrence after liver transplantation. Surg. Oncol. 2016, 25, 419–428. [Google Scholar] [CrossRef]
- Cruz, F.D.; Matushansky, I. Solid tumor differentiation therapy-is it possible? Oncotarget 2012, 3, 559–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Jin, X.; Kim, H. Cancer stem cells and differentiation therapy. Tumour Biol. 2017, 39, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, F.; Nagl, F.; Mazur, P.K.; Lee, M.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Schmid, R.M.; Siveke, J.T. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology 2008, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Gil-Garcia, B.; Baladron, V. The complex role of NOTCH receptors and their ligands in the development of hepatoblastoma, cholangiocarcinoma and hepatocellular carcinoma. Biol. Cell 2016, 108, 29–40. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Li, J.; Zheng, J.; Wei, A. The Carcinogenic Role of the Notch Signaling Pathway in the Development of Hepatocellular Carcinoma. J. Cancer 2019, 10, 1570–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, R.; An, H.; Yu, Y.; Zhang, M.; Liu, S.; Xu, H.; Guo, Z.; Cheng, T.; Cao, X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003, 63, 8323–8329. [Google Scholar] [PubMed]
- Viatour, P.; Ehmer, U.; Saddic, L.A.; Dorrell, C.; Andersen, J.B.; Lin, C.; Zmoos, A.F.; Mazur, P.K.; Schaffer, B.E.; Ostermeier, A.; et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J. Exp. Med. 2011, 208, 1963–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Rodriguez-Carunchio, L.; Sole, M.; Thung, S.; Stanger, B.Z.; et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology 2012, 143, 1660–1669.e1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill, M.T.; Tornillo, L.; Fritzius, T.; Terracciano, L.; Semela, D.; Bettler, B.; Heim, M.H.; Tchorz, J.S. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 2013, 57, 1607–1619. [Google Scholar] [CrossRef]
- Zhu, C.; Ho, Y.J.; Salomao, M.A.; Dapito, D.H.; Bartolome, A.; Schwabe, R.F.; Lee, J.S.; Lowe, S.W.; Pajvani, U.B. Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J. Hepatol. 2021, 74, 613–626. [Google Scholar] [CrossRef]
- Sekiya, S.; Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Investig. 2012, 122, 3914–3918. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dong, M.; Xu, Z.; Song, X.; Zhang, S.; Qiao, Y.; Che, L.; Gordan, J.; Hu, K.; Liu, Y.; et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018, 37, 3229–3242. [Google Scholar] [CrossRef]
- Nakano, Y.; Nakao, S.; Sueoka, M.; Kasahara, D.; Tanno, Y.; Sumiyoshi, H.; Itoh, T.; Miyajima, A.; Hozumi, K.; Inagaki, Y. Two distinct Notch signals, Delta-like 4/Notch1 and Jagged-1/Notch2, antagonistically regulate chemical hepatocarcinogenesis in mice. Commun. Biol. 2022, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, P.J.; Oderberg, I.M.; Goessling, W. There Is Something Fishy About Liver Cancer: Zebrafish Models of Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 347–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Emelyanov, A.; Koh, C.H.; Spitsbergen, J.M.; Parinov, S.; Gong, Z. An inducible kras(V12) transgenic zebrafish model for liver tumorigenesis and chemical drug screening. Dis. Model. Mech. 2012, 5, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Petitt, M.; Gamboa, M.; Huang, M.; Dhal, S.; Druzin, M.L.; Wu, J.C.; Chen-Tsai, Y.; Nayak, N.R. Transient, inducible, placenta-specific gene expression in mice. Endocrinology 2012, 153, 5637–5644. [Google Scholar] [CrossRef] [Green Version]
- Reboredo, M.; Kramer, M.G.; Smerdou, C.; Prieto, J.; De Las Rivas, J. Transcriptomic effects of Tet-on and mifepristone-inducible systems in mouse liver. Hum. Gene Ther. 2008, 19, 1233–1247. [Google Scholar] [CrossRef] [Green Version]
- Seelinger, M.; Otterlei, M. Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int. J. Mol. Sci. 2020, 21, 693. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Lu, H.; Zou, Q.; Luo, L. Regeneration of liver after extreme hepatocyte loss occurs mainly via biliary transdifferentiation in zebrafish. Gastroenterology 2014, 146, 789–800.e8. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Golubkov, V.S.; Han, W.; Correa, R.G.; Zhou, Y.; Lee, S.; Strongin, A.Y.; Dong, P.D. Identification of Annexin A4 as a hepatopancreas factor involved in liver cell survival. Dev. Biol. 2014, 395, 96–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.L.; Zhou, Z.Y.; Ruan, B.; Fang, Z.Q.; Ding, J.; Liu, J.J.; Song, P.; Xu, H.; Xu, C.; Yue, Z.S.; et al. Notch-Regulated c-Kit Positive Liver Sinusoidal Endothelial Cells Contribute to Liver Zonation and Regeneration. Cell. Mol. Gastroenterol. Hepatol. 2022, in press. [CrossRef] [PubMed]
- Weng, A.P.; Nam, Y.; Wolfe, M.S.; Pear, W.S.; Griffin, J.D.; Blacklow, S.C.; Aster, J.C. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol. Cell. Biol. 2003, 23, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Borikova, A.L.; Ben-Yair, R.; Guner-Ataman, B.; MacRae, C.A.; Lee, R.T.; Burns, C.G.; Burns, C.E. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 1403–1408. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, X.; Ni, R.; Chen, Q.; Yang, Q.; He, J.; Luo, L. Acute brain vascular regeneration occurs via lymphatic transdifferentiation. Dev. Cell 2021, 56, 3115–3127.e6. [Google Scholar] [CrossRef]
- Tanimizu, N.; Nishikawa, Y.; Ichinohe, N.; Akiyama, H.; Mitaka, T. Sry HMG box protein 9-positive (Sox9+) epithelial cell adhesion molecule-negative (EpCAM-) biphenotypic cells derived from hepatocytes are involved in mouse liver regeneration. J. Biol. Chem. 2014, 289, 7589–7598. [Google Scholar] [CrossRef] [Green Version]
- Panda, M.; Tripathi, S.K.; Biswal, B.K. SOX9: An emerging driving factor from cancer progression to drug resistance. Biochim. Biophys Acta Rev. Cancer 2021, 1875, 188517. [Google Scholar] [CrossRef]
- Song, S.; Maru, D.M.; Ajani, J.A.; Chan, C.H.; Honjo, S.; Lin, H.K.; Correa, A.; Hofstetter, W.L.; Davila, M.; Stroehlein, J.; et al. Loss of TGF-beta adaptor beta2SP activates notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res. 2013, 73, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Delous, M.; Yin, C.; Shin, D.; Ninov, N.; Debrito Carten, J.; Pan, L.; Ma, T.P.; Farber, S.A.; Moens, C.B.; Stainier, D.Y. Sox9b is a key regulator of pancreaticobiliary ductal system development. PLoS Genet 2012, 8, e1002754. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Sodji, Q.H.; Oyelere, A.K. Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers 2022, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- de Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Chao, J.; Zhao, S.; Sun, H. Dedifferentiation of hepatocellular carcinoma: Molecular mechanisms and therapeutic implications. Am. J. Transl. Res. 2020, 12, 2099–2109. [Google Scholar]
- Zhou, T.; Li, S.; Xiang, D.; Liu, J.; Sun, W.; Cui, X.; Ning, B.; Li, X.; Cheng, Z.; Jiang, W.; et al. m6A RNA methylation-mediated HNF3gamma reduction renders hepatocellular carcinoma dedifferentiation and sorafenib resistance. Signal Transduct. Target.Ther. 2020, 5, 296. [Google Scholar] [CrossRef] [PubMed]
- Risom, T.; Langer, E.M.; Chapman, M.P.; Rantala, J.; Fields, A.J.; Boniface, C.; Alvarez, M.J.; Kendsersky, N.D.; Pelz, C.R.; Johnson-Camacho, K.; et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 2018, 9, 3815. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Medina, M.; Avendano-Felix, M.; Lizarraga-Verdugo, E.; Bermudez, M.; Romero-Quintana, J.G.; Ramos-Payan, R.; Ruiz-Garcia, E.; Lopez-Camarillo, C. SOX9 Stem-Cell Factor: Clinical and Functional Relevance in Cancer. J. Oncol. 2019, 2019, 6754040. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, Z.; Jiang, B.; Peng, R.; Ma, Z.; Lu, J. SOX9 Overexpression Promotes Glioma Metastasis via Wnt/beta-Catenin Signaling. Cell Biochem. Biophys. 2015, 73, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.L.; Back, J.H.; Chaudhary, S.C.; Zhu, Y.; Athar, M.; Bickers, D.R. SOX9 Transcriptionally Regulates mTOR-Induced Proliferation of Basal Cell Carcinomas. J. Investig. Dermatol. 2018, 138, 1716–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Chen, J.; Wei, X.; Leng, H.; Mu, H.; Cai, P.; Luo, L. Mammalian Target of Rapamycin Complex 1 Signaling Is Required for the Dedifferentiation From Biliary Cell to Bipotential Progenitor Cell in Zebrafish Liver Regeneration. Hepatology 2019, 70, 2092–2106. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Bauer, J.; Wise, P.; Kruger, M.; Simonsen, U.; Wehland, M.; Infanger, M.; Corydon, T.J. The role of SOX family members in solid tumours and metastasis. Semin. Cancer Biol. 2020, 67, 122–153. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Fang, S.; Liu, M.; Li, L.; Zhang, F.F.; Li, Y.; Yan, Q.; Cui, Y.Z.; Zhu, Y.H.; Yuan, Y.F.; Guan, X.Y. Lymphoid enhancer-binding factor-1 promotes stemness and poor differentiation of hepatocellular carcinoma by directly activating the NOTCH pathway. Oncogene 2019, 38, 4061–4074. [Google Scholar] [CrossRef]
- He, J.; Mo, D.; Chen, J.; Luo, L. Combined whole-mount fluorescence in situ hybridization and antibody staining in zebrafish embryos and larvae. Nat. Protoc. 2020, 15, 3361–3379. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; He, J.; Shi, W.; Jiang, Z.; Gao, L.; Jiang, Y.; Ni, R.; Yang, Q.; Luo, L. A single-cell-resolution fate map of endoderm reveals demarcation of pancreatic progenitors by cell cycle. Proc. Natl. Acad. Sci. USA 2021, 118, e2025793118. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; He, J.; Ni, R.; Yang, Q.; Zhang, Y.; Luo, L. Cerebrovascular Injuries Induce Lymphatic Invasion into Brain Parenchyma to Guide Vascular Regeneration in Zebrafish. Dev. Cell 2019, 49, 697–710.e5. [Google Scholar] [CrossRef]
- Liu, C.; Wu, C.; Yang, Q.; Gao, J.; Li, L.; Yang, D.; Luo, L. Macrophages Mediate the Repair of Brain Vascular Rupture through Direct Physical Adhesion and Mechanical Traction. Immunity 2016, 44, 1162–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Ma, J.; Yang, Y.; Shi, W.; Luo, L. EpCAM is an endoderm-specific Wnt derepressor that licenses hepatic development. Dev. Cell 2013, 24, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, P.; Mao, X.; Zhao, J.; Nie, L.; Jiang, Y.; Yang, Q.; Ni, R.; He, J.; Luo, L. Farnesoid X Receptor is Required for the Redifferentiation of Bi-potential Progenitor Cells during Biliary-Mediated Zebrafish Liver Regeneration. Hepatology 2021, 74, 3345–3361. [Google Scholar] [CrossRef]
- van Putten, M.; de Winter, C.; van Roon-Mom, W.; van Ommen, G.J.; AC’t Hoen, P.; Aartsma-Rus, A. A 3 months mild functional test regime does not affect disease parameters in young mdx mice. Neuromuscul. Disord. 2010, 20, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Rangan, G.K.; Tesch, G.H. Quantification of renal pathology by image analysis. Nephrology 2007, 12, 553–558. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Chen, Q.; Ma, J. Notch–Sox9 Axis Mediates Hepatocyte Dedifferentiation in KrasG12V-Induced Zebrafish Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 4705. https://doi.org/10.3390/ijms23094705
Sun J, Chen Q, Ma J. Notch–Sox9 Axis Mediates Hepatocyte Dedifferentiation in KrasG12V-Induced Zebrafish Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2022; 23(9):4705. https://doi.org/10.3390/ijms23094705
Chicago/Turabian StyleSun, Junhui, Qi Chen, and Jianlong Ma. 2022. "Notch–Sox9 Axis Mediates Hepatocyte Dedifferentiation in KrasG12V-Induced Zebrafish Hepatocellular Carcinoma" International Journal of Molecular Sciences 23, no. 9: 4705. https://doi.org/10.3390/ijms23094705