Abstract
Sclerostin, a glycoprotein encoded by the SOST gene, is mainly produced by mature osteocytes and is a critical regulator of bone formation through its inhibitory effect on Wnt signaling. Osteocytes are differentiated osteoblasts that form a vast and highly complex communication network and orchestrate osteogenesis in response to both mechanical and hormonal cues. The three most commonly described pathways of SOST gene regulation are mechanotransduction, Wnt/β-catenin, and steroid signaling. Downregulation of SOST and thereby upregulation of local Wnt signaling is required for the osteogenic response to mechanical loading. This review covers recent findings concerning the identification of SOST, in vitro regulation of SOST gene expression, structural and functional properties of sclerostin, pathophysiology, biological variability, and recent assay developments for measuring circulating sclerostin. The three-dimensional structure of human sclerostin was generated with the AlphaFold Protein Structure Database applying a novel deep learning algorithm based on the amino acid sequence. The functional properties of the 3-loop conformation within the tertiary structure of sclerostin and molecular interaction with low-density lipoprotein receptor-related protein 6 (LRP6) are also reviewed. Second-generation immunoassays for intact/biointact sclerostin have recently been developed, which might overcome some of the reported methodological obstacles. Sclerostin assay standardization would be a long-term objective to overcome some of the problems with assay discrepancies. Besides the use of age- and sex-specific reference intervals for sclerostin, it is also pivotal to use assay-specific reference intervals since available immunoassays vary widely in their methodological characteristics.
Keywords:
β-catenin; bone; bone formation; immunoassay; LRP6; mechanotransduction; osteocyte; reference interval; SOST; Wnt signaling 1. Introduction
In 1637, the island of Urk (now connected to the mainland), in the northern part of the Netherlands, was ravaged by the plague, killing off half of the island’s 300 inhabitants. This event, with its subsequent reduction in the potential number of romantic suitors, would eventually lead to the marriage and inbreeding of a particular Urk-dwelling couple in 1751 [1]. Their descendants, and the distinctive bone disorder they passed along through generations, led van Buchem et al. to the discovery and portrayal of what they named hyperostosis corticalis generalisata familiaris in 1955 [2]. A characteristic phenotypic feature was generalized skeletal sclerosis leading to increased bone mineral density typically affecting the cranial base, jaw, and diaphyseal cortex of long bones. The cranial hyperostosis was particularly troublesome since it caused the narrowing of cranial neuroforamina, which subsequently led to facial palsy and deafness while also causing increased intracranial pressure in early adulthood. In 1958, Truswell described a similar disorder found in the Afrikaner community of South Africa, which he named osteopetrosis with syndactyly [3]. Those affected displayed virtually identical cranial features as in the disease described by van Buchem et al. [2], but these individuals were typically of tall stature and often had signs of syndactyly (fused digits).
In view of the ancestral links between the Dutch and the Afrikaners, a speculation arose that the two conditions might result from mutations in the same gene and that those afflicted might have a common ancestry [4]. In 1998, genome-wide studies were performed on a group of Dutch individuals and their families, which led to the mapping of van Buchem disease to chromosome 17q12-q21 [1]. The mutation has been described as autosomal recessive with loss of function. A year later linkage analysis performed in two inbred families with sclerosteosis proved that the common faulty gene was indeed the same. They further proposed that the possible function of the gene is linked to either inhibition of bone formation or stimulation of bone resorption [1]. The genetic lesions responsible for each disease, however, were shown to have distinct locations, thus disproving the earlier speculation that the two disorders represented a common ancestry.
Two groups of researchers independently identified the SOST gene in 2001 [5,6]. This gene was shown to encode a protein with a signal peptide for secretion and a cysteine-knot motif. Loss-of-function mutations in this gene were found in patients with sclerosteosis and thus the physiological role of the SOST protein was hypothesized to be suppression of bone formation. Sequencing analysis in two distinctive families from Brazil and USA revealed different mutations in exon 2 that caused a premature stop codon [5,7]. A 52 kb deletion, located 35 kb downstream of SOST, was identified as the culprit of van Buchem disease [8]. Later, this deletion was shown to be a noncoding enhancer element, required for the postnatal expression of the SOST gene product sclerostin [9].
Although the majority of sclerosteosis cases have been found in the Afrikaner community of South Africa that are descendants of the Dutch settlers from the 17th century, several cases from the USA, Brazil, Germany, Morocco, Turkey, Saudi Arabia, Egypt, Senegal, India, Japan, and China have been reported with 11 different mutations [10]. In addition, two mutations in low-density lipoprotein receptor-related protein 4 (LRP4) have been found in sclerosteosis patients, independent of the sclerostin genotype, demonstrating that LRP4 interacts with sclerostin or mediates its function [11,12,13].
Several other genetic variants have recently been described with various effects on SOST expression or gene regulation. The most frequent genetic variant, rs17882143 (SOST-p. Val10Ile), located in the signaling peptide, is associated with decreased translocation of sclerostin intra- and extracellularly. It may be associated with a weak decrease in SOST expression and the development of non-pathogenic high bone mineral density in the general population [14]. Another variant located in the TATA box motif caused decreased promoter activity [14]. However, information is scarce about the distribution of rare genetic and non-pathological SOST variants in the general population and their role in skeletal pathogenesis [15].
2. Osteogenesis and Regulation of SOST Expression
Osteocytes represent 90–95% of all bone cells in the adult skeleton and are believed to have a lifespan of up to several decades, while osteoclasts live only for a few days and osteoblasts for a few months [16,17,18]. It is believed that the precursors of osteoblasts are skeletal stem cells, formerly known as multipotent mesenchymal stem cells, which also give rise to chondrocytes, muscle cells, and adipocytes [19,20,21].
Bone remodeling is the process by which the body maintains healthy bones and regulates mineral homeostasis. Osteoclasts resorb old bone tissue and osteoblasts form new bone by a mechanism that is strictly regulated by close interaction, known as coupling, of both types of cells. The whole process takes place in small areas of the cortical bone or the trabecular surface, called basic multicellular units, which are formed by osteoclasts, osteoblasts, osteocytes, bone lining cells, and blood vessels [16,22,23]. This complex process of bone remodeling is regulated by several interacting factors including genetic, mechanical (loading, exercise), vascular/nerve, nutritional and by various autocrine, paracrine, and endocrine signals.
Sclerostin was first identified in adult human osteocytes [24], and it has recently been shown that SOST is one of the nine most enriched genes in the osteocyte transcriptome signature (SOST, MEPE, NGEF, WNT1, ACKR3, TGFB2, SEMA3E, IRX5, and DMP1) [25]. The strong expression of SOST in osteocytes implies a central role in the regulation of bone homeostasis [26]. Although osteocytes are the main cellular source of sclerostin, SOST RNA is also expressed in other human and mouse tissues such as cartilage, kidney, heart, liver, epididymis, and vas deferens of the testis, pyloric sphincter, carotid arteries, and parts of the central nervous system [5,27,28]. The biological function of sclerostin outside of the skeleton is less well known. Overexpression of SOST in transgenic mice results in reduced osteoblast activity and a lower degree of bone formation resulting in low bone mass and reduced bone strength [26].
SOST contains two exons and one intron. The expression is regulated by the non-coding evolutionarily conserved region 5 (ECR5) enhancer site located 35 kb downstream of SOST and by a proximal promoter upstream. The transcriptional regulation through either the enhancer or promoter is pathway dependent [29]. The three most commonly described pathways of SOST regulation in osteocytes, further discussed in this review, are mechanotransduction [30], Wnt/β-catenin, and steroid signaling [31,32].
2.1. SOST Regulation by Mechanotransduction
Mechanical loading is a pivotal factor for bone remodeling since microgravity has a negative impact on bone health in space [33]. In addition, recent research on rodents in space has demonstrated increased osteoclast activity, altered osteoblast differentiation, and osteocyte function [34]. Mice acquired, during a 30-day-long spaceflight, a dramatic increase in SOST mRNA expression [35]. Despite the limited research reported (due to practical reasons) on the effect of microgravity on bone health, this pointed to the hypothesis that mechanical loading is an important factor for maintaining a balanced bone remodeling and that this effect could be regulated by sclerostin. Mechanical stimulation by whole-body vibration, a non-pharmacological approach to increase bone mass, was originally developed to prevent loss of bone and muscle mass during prolonged spaceflights [36]. It has also been shown clinically that whole-body vibration results in reduced serum levels of sclerostin, which implies a direct effect on osteocytes through mechanical stimulation [37,38].
The main mechanosensory cell in the skeleton is the osteocyte. Several in vitro and in vivo studies have described molecular pathways in osteocytes that link mechanotransduction to SOST regulation [39]. Studying the expression of SOST with in vivo models is complex and depends on multiple factors, while the isolation and culturing of primary osteocytes is technically challenging [40]. Therefore, human osteocyte-like cell lines (e.g., MLO-Y4, Ocy 454, and OMGFP66) and/or differentiated osteoblasts and mesenchymal stem cells are mainly used [41,42,43,44,45,46,47].
Low rates of fluid flow shear stress (FFSS) are sensed from the extracellular matrix (ECM) by αVβ3 integrins on the dendrites of osteocytes which, through phosphoinositide 3-kinase (PI3K-Akt), activate the α5β1 integrin-coupled opening of connexin 43 (Cx43)-gap junction channels in osteocytes. Cx43 opening leads to prostaglandin E2 (PGE2) efflux and activation of the PGE2 receptor subtypes EP2 and EP4, which downregulates SOST through β-catenin activation [41,48,49]. FFSS also stimulates Ca-channel opening (TRPV4 and Piezzo1) in the cellular body and Ca2+ influx, which also activates Akt [42,43,50]. These findings suggest that the Akt pathway is one of the major intracellular mediators of mechanical stimuli in osteocytes that leads to down-regulation of SOST (Figure 1).
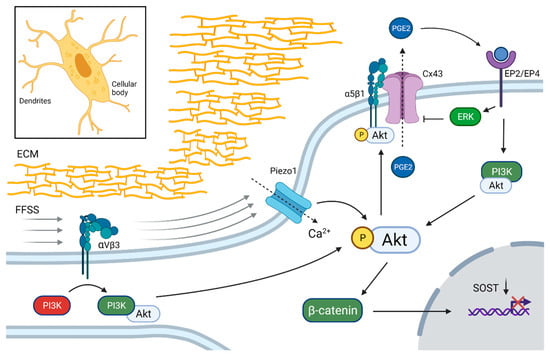
Figure 1.
Down-regulation of SOST in osteocytes by mechanotransduction. Integrin αVβ3 in dendritic processes sense fluid flow shear stress (FFSS) applied on the extracellular matrix (ECM). The subsequent activation of phosphoinositide 3-kinase (PI3K) phosphorylates Akt, which activates integrin-mediated connexin 43 (Cx43) channel opening on the cellular body. Prostaglandin E2 (PGE2) is transported out of the osteocyte and binds to EP2 and EP4 receptors, which further activates the Akt and extracellular signal-regulated kinase (ERK) pathways and β-catenin, which down-regulates SOST expression. FFSS activates also Ca2+ influx through Piezo1 channels, which enables enhanced Akt activation. Black dotted arrows indicate transport through membrane channels. Created with BioRender.com, accessed on 15 February 2022.
Another mechanotransduction pathway has been proposed by Wein et al. [44]. They found that histone deacetylase 4/5 (HDAC4/5) regulates SOST expression by histone acetylation at the ECR5 enhancer site, which prevents myocyte-specific enhancer factor 2C (MEF2C) from binding. The transcription factor MEF2C is needed for SOST expression and both acetylation at the ECR5 enhancer site and MEF2C knock-down or deletion caused decreased SOST expression and also increased bone mass in mice [44,51,52]. In a subsequent study, Sato et al. [45] demonstrated that HDAC4/5 was phosphorylated by focal adhesion kinase and translocated to the nucleus upon FFSS-induced integrin dissociation. Furthermore, lack of FFSS and HDAC4/5 knock-down leads to MEF2C binding at the ECR5 site and increased SOST expression, suggesting that phosphorylated HDAC4/5 is a negative regulator of SOST (Figure 2) [45,51].
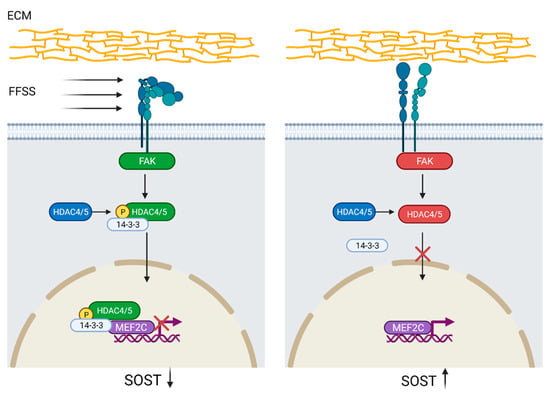
Figure 2.
Regulation of SOST by histone deacetylase 4/5 (HDAC4/5) activation. Integrin-disintegration, due to fluid flow shear stress (FFSS) in the extracellular matrix (ECM), activates focal adhesion kinase (FAK), which subsequently phosphorylates HDAC4/5. Phosphorylated HDAC4/5 binds to protein 14-3-3 leading to nuclear translocation where, by binding to myocyte-specific enhancer factor 2C (MEF2C) and histone deacetylation at the evolutionarily conserved region 5 (ECR5) site, prevents SOST expression. Lack of FFSS precludes HDAC4/5 phosphorylation and thus nuclear translocation, which promotes MEF2C binding to ECR5 and SOST upregulation. SOST downregulation = black arrow down; SOST upregulation = black arrow up. Created with BioRender.com, accessed on 15 February 2022.
2.2. SOST Regulation by Steroid Hormones
Apart from mechanotransduction, the expression of SOST is also regulated by steroid hormone signaling such as estrogen receptors (ER) (Figure 3). The two receptors ERα and ERβ have opposing effects on SOST expression. It has been also shown that estradiol downregulates SOST expression. While the downregulation by estradiol is mediated by binding to ERβ, the ERα ligand independently maintains SOST expression. ERα and ERβ compete for the same SOST regulatory site and estradiol enhances the binding of ERβ and thereby prevents ERα from binding to the gene. Loss or inhibition of ERα results also in down-regulation of basal SOST expression [53]. Furthermore, treatment with estradiol in human mesenchymal stem cells activates the Wnt/ERα/β-catenin signaling pathway, which results in increased levels of total and active β-catenin [31]. Mechanical strain performed on ulnae from ovariectomized mice resulted in decreased β-catenin activation and increased expression of SOST in comparison with sham mice, which suggests that an estrogen/Wnt/β-catenin pathway crosstalk is involved in the regulation of SOST [32].
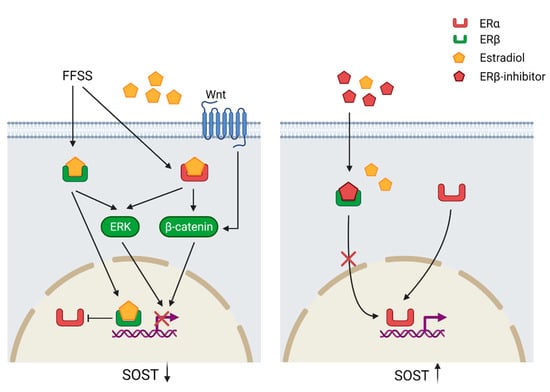
Figure 3.
Regulation of SOST through the estrogen receptor (ER) pathway. Fluid flow shear stress (FFSS) and estradiol (E2) binding to ERβ suppresses ERα binding to the regulatory motif of SOST resulting in SOST down-regulation. Estradiol binding to the ERs triggers the extracellular signal-regulated kinase (ERK) and Wnt/β-catenin, which suppress SOST expression. The ERα ligand independently maintains basal SOST expression and Erβ inhibition prevents down-regulation of SOST. SOST downregulation = black arrow down; SOST upregulation = black arrow up. Created with BioRender.com, accessed on 15 February 2022.
Wijenayaka et al. [54] demonstrated that the steroid hormone 1,25-dihydroxyvitamin D (1,25(OH)2D) regulates SOST expression. 1,25(OH)2D induced expression of SOST in osteoblast-like SaOS-2 cells by binding to a vitamin D response element upstream to SOST but not by binding to the enhancer site ECR5 [54,55]. Although these findings suggest a direct action of 1,25(OH)2D on the regulation of SOST, additional studies are needed to elucidate the precise mechanism of action and the regulatory effect in different osteoblast and osteocyte-like cell lines.
3. Structural Properties of Human Sclerostin
The product of SOST, sclerostin, is a 24 kDa and 213 amino acid-long glycoprotein with a secretion signaling peptide comprising the first 23 amino acids [5]. The circulating form of sclerostin is 22 kDa after cleavage of the signal peptide. Nuclear magnetic resonance analysis shows that the tertiary structure consists of three loops, a cysteine-knot motif with three disulfide bonds, and N- and C-terminal spacer arms [56]. Loops 1 and 3 contain two anti-parallel β-sheets that link with a disulfide bond on the top. Whereas loops 1 and 3 form stable structures, loop 2 is more flexible adopting different stable conformations upon ligand interactions. A structure-based sequence alignment of sclerostin with proteins from the cysteine-knot family reveals a highly conserved cysteine-knot motif, buried in the core of the protein, stabilizing its structure. Most other cysteine-knot proteins form functional homo- or heterodimers. To our knowledge, there is only one experimental study that suggests a possible dimerization or trimerization of sclerostin [57]; however, these findings need to be confirmed in future studies. Moreover, in contrast to the molecular structure of sclerostin, none of the previously identified structural homologs from the cysteine-knot family have long flexible N- and C-terminal arms [56].
We performed a computational analysis of the tertiary structure of human sclerostin in the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/entry/Q9BQB4) (accessed on 19 September 2021) and the three-dimensional structure of sclerostin is depicted in Figure 4. AlphaFold is a novel deep learning algorithm developed by DeepMind (London, UK) that performs protein folding predictions based on the amino acid sequence and provides accuracy calculations of the predicted three-dimensional structure [58,59,60]. AlphaFold provides a per-residue confidence measurement from 0 to 100 of the model termed the predicted local-distance difference test (pLDDT). The AlphaFold structure prediction of human sclerostin confirmed the previously described 3-loop conformation with high to very high model confidence (70 > pLDDT > 90; pLDDT > 90; light blue, dark blue) of loop 1 and 3 (Figure 4A). As mentioned, loops 1 and 3 have stable structures and consist of hydrophobic amino acids with the hydrophobic side buried in the core of the loop structure. Additionally, the cysteine-knot motif with three disulfide bonds in close proximity to each other (confidence pLDDT score between 80 and 90) further describes the stable core structure of sclerostin. Furthermore, the predicted aligned error (Figure 4B), that is the expected position error between the aligned residues in Ångströms, is low for the core structure, defining four inner-sequence domains (β-sheets for loops 1 and 3) with high proximity of the residues in the predicted core structure. However, loop 2 and the two spacer arms exhibit a three-dimensional structure with low confidence (70 > pLDDT > 50; pLDDT < 50; yellow, orange) and residues with low proximity. It is worth mentioning that AlphaFold only provides a predicted structure and from the low confidence score for loop 2 and the spacer arms it can be deduced that the three-dimensional orientation, proximity, or correct folding, would circumstantially vary depending on different physicochemical conditions and molecular interactions.
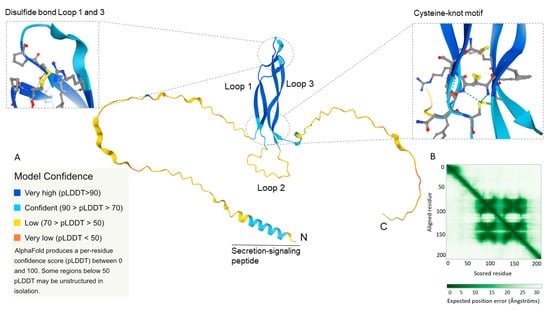
Figure 4.
The three-dimensional structure of human sclerostin was generated from the amino acid sequence in the novel AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/entry/Q9BQB4) (accessed on 19 September 2021) [58]. The protein comprises a core structure of loops 1 and 3, each having two anti-parallel running β-sheets linking with three disulfide bonds (cysteine-knot motif, magnification to the right; disulfide bond in yellow) and one disulfide bond in the top (magnification to the left; disulfide bond in yellow). Sclerostin contains also a flexible second loop in the bases and two N- and C-terminal spacer arms. (A) Confidence score (0–100) of model accuracy based on per-residue predicted local-distance difference test (pLDDT). Loops 1 and 3 have a high to very high confidence score (pLDDT > 70) and loop 2 and the spacer arms low confidence score (pLDDT < 70). (B) Expected position error, showing the possible interaction/proximity between the residues, depending on the distance between each residue (in Ångströms). Sclerostin has four closely aligned inner-sequence core domains, each containing one β-sheet.
Sclerostin interacts with glycosaminoglycans such as heparin/heparan sulfate by electrostatic interactions between arginine and lysine side chains on the hydrophilic surface and sulfate groups of heparin. This molecular interaction mediates the transportation of sclerostin to the cell surface and embedding in the extracellular matrix [56]. Heparan sulfate is produced by bone cells and is a component of the extracellular matrix. It also facilitates the excretion of soluble proteins from the cells. Sclerostin interacts preferably with full-length heparin and this interaction is enhanced by sulfation and high divalent cation concentrations such as Ca2+ and Zn2+ [61]. Sclerostin interacts also with the co-receptors LRP4, 5, and 6 and thereby impedes the binding of mainly Wnt1 but also Wnt3a. It has been reported that sclerostin is a more potent inhibitor of Wnt1 in comparison with Wnt3a and also that the binding mechanism of sclerostin to the Wnt1 and Wnt3a receptors differs [62]. Through crystal structure analysis, Kim and colleagues [63] proposed an interactive mechanism of sclerostin with LRP6 (Figure 5). LRP6 is a transmembrane protein that consists of four identical ectodomains (E1, E2, E3, and E4), and the co-receptor interaction of LRP6 with the G-protein coupled receptor Frizzled depends on the type of ligand binding. While Wnt3a interacts with E3 and E4, and another Wnt-inhibitor dickkopf-1 (DKK1) interacts with the E1 and E4 ectodomains, both Wnt1 and sclerostin interact with the E1 and E2 ectodomains of LRP6 [62]. Sclerostin interacts with E1 and E2 by binding with loop 2 and partially with the C-terminal arm, respectively (Figure 5) [63]. This interaction inhibits directly the binding of Wnt1 and allosterically prevents the binding of Wnt3a to LRP6 and thereby prevents receptor-coupling with Frizzled on osteoblasts [62]. This inhibition leads to extracellular events that will be further discussed in this review.
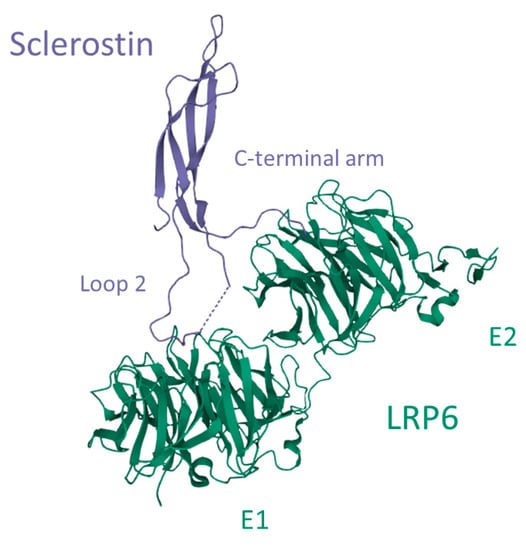
Figure 5.
Interaction between sclerostin and the low-density lipoprotein receptor-related protein 6 (LRP6). Loop 2 of sclerostin binds to the E1 ectodomain of LRP6, while a part of the C-terminal arm interacts with the E2 ectodomain. Image created in the Research Collaboratory for Structural Bioinformatics—Protein Data Bank with the Mol* Viewer [64], based on the crystal structure LRP6 E1-E2-SOST complex, PDB-ID: 6L6R [63].
4. Functional Properties of Sclerostin
The Wnt signaling pathway is a critical regulator of the development and maintenance of tissues, including bone, working through the stimulation of Runx2 gene expression [65,66]. The discovery of the Wnt/β-catenin signaling system in bone led to an increased interest in Wnt signaling within the bone research community [67,68]. Canonical Wnt signaling promotes osteogenesis through its effects on mesenchymal stem cells by promoting osteoblastic lineage whilst inhibiting osteoclast, adipocyte, and chondrocyte differentiation [67,69,70]. Binding of Wnt to its receptor complex, LRP5/6, and the Frizzled receptor, facilitates β-catenin stabilization and accumulation in the cytoplasm. β-catenin is translocated to the nucleus, where it binds to T-cell factor/lymphoid enhancer factor (TCF/LEF), causing displacement of transcriptional corepressors, and initiates transcription of Wnt target genes [71]. Sclerostin is a negative regulator of Wnt signaling [24,37]. By binding to LPR4 and LPR5/6, sclerostin inhibits canonical Wnt signaling leading to phosphorylation of β-catenin and thereby enhances its degradation by proteasomes (Figure 6) [72]. DKK1 is another endogenously secreted factor that acts as an inhibitor of the Wnt pathway through the Wnt co-receptor LPR5/6 [73]. A potential role in Wnt signaling has also been suggested for fibroblast growth factor 23 (a regulator of phosphate metabolism secreted by osteoblasts and osteocytes). Mäkitie et al. [74] found normal levels of sclerostin and DKK1 but increased levels of fibroblast growth factor 23 in WNT1 mutation-positive individuals with impaired Wnt signaling and early-onset osteoporosis.
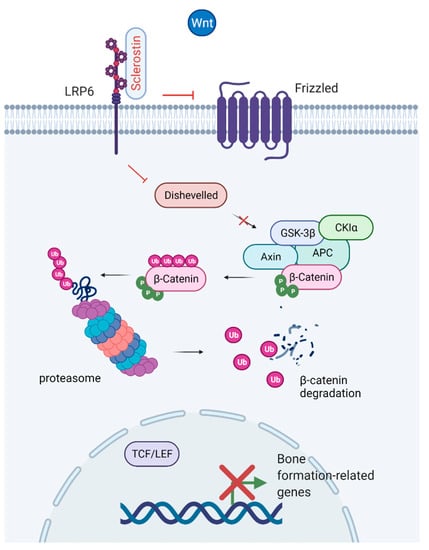
Figure 6.
Inhibition of the Wnt/β-catenin canonical pathway. Binding of sclerostin to LRP6 prevents coupling with the Frizzled receptor. Deactivation of the receptor results in phosphorylation of β-catenin by the APC-GSK3β complex, translocation of β-catenin to the proteasome and degradation. The enhanced degradation of β-catenin prevents nuclear translocation and binding to the T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor complex, which leads to reduced expression of genes related to bone formation. Created with BioRender.com, accessed on 15 February 2022.
5. Pathophysiology and Biological Variability of Sclerostin
Inheritance, environmental factors, age, sex, body mass index, and total body fat content all influence the circulating levels of sclerostin. Declining bone mass with age is associated with increased expression of sclerostin in osteocytes with a subsequent reduction in Wnt/β-catenin signaling in bone cells [75,76,77]. Unlike the bone resorption marker C-terminal telopeptide of type I collagen (CTX) and the bone formation marker osteocalcin, serum sclerostin is unaffected by the time of blood sampling [78,79].
Different pathological disorders may influence the expression of SOST and subsequently the circulating levels of sclerostin. Paget’s disease of bone (PDB) is a focal chronic metabolic disorder affecting mostly elderly individuals. PDB is characterized by unbalanced bone remodeling resulting in areas of increased bone resorption and other areas with abnormal formation of new bone, with the primary cellular abnormality in osteoclasts [80]. Elevated sclerostin levels were observed in older patients with PDB although they did not correlate with other markers of bone turnover [81]. Yavropoulou et al. showed that circulating sclerostin levels are significantly increased in patients with increased bone turnover, regardless of underlying pathologies such as PDB [82]. Another study did not find any differences in sclerostin levels between patients with active or inactive PDB [83].
Patients with chronic kidney disease have higher levels of sclerostin in comparison with healthy individuals and serum sclerostin tends to increase as the kidney function declines. In reported mice models, an early increase in sclerostin-positive osteocytes correlated with an increased expression of inactive phosphorylated β-catenin, which suggests an overall repression in Wnt/β-catenin signaling that may contribute to the progression of chronic kidney disease–mineral bone disorder (a.k.a. CKD–MBD) [84,85]. In humans, sclerostin was reported to be positively correlated with age and to be higher in men than women even in patients with chronic kidney disease [86,87,88,89]. Boltenstål and colleagues [90] studied a cohort of patients with chronic kidney disease stages 3–5D and showed that sclerostin levels were higher in low bone turnover patients in comparison with non-low bone turnover patients. A recent study by Nakagawa and colleagues on 654 hemodialysis patients showed that higher levels of sclerostin were associated with lower levels of parathyroid hormone and bone-specific alkaline phosphatase [91]. However, these associations were substantially attenuated when the results were statistically adjusted for parathyroid hormone, which suggests that sclerostin is not the main mediator driving the effects of parathyroid hormone on bone remodeling.
Diabetes is associated with alterations in bone mineral density, growth, and bone remodeling, with an increased risk of fractures [92,93,94]. Bone-derived factors including osteocalcin, osteoprotegerin, and sclerostin may be altered because of disruption of the glucose metabolism [95], and different mechanisms have been suggested in the pathogenesis of diabetes-associated skeletal disease [96]. A recent review anticipated the possible interaction between bone and fat tissue in the regulation of glucose metabolism [97]. Sclerostin was negatively associated with C-peptide and HbA1c in children with type 1 diabetes implying an interaction between sclerostin and glucose metabolism [98]. Elevated levels of sclerostin, independent of age and sex, have also been shown in patients with type 2 diabetes [99,100,101].
Sclerostin as a Treatment Target
Osteoporosis is a global health problem and the most common chronic metabolic disease with a higher incidence in Caucasians, older individuals, and women. The European Foundation for Osteoporosis and Bone Disease defined osteoporosis as a progressive systemic skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase in bone fragility and susceptibility to fracture [102]. Biochemical markers of bone turnover are clinically useful for monitoring the treatment of osteoporosis, although they cannot be used for the diagnosis of osteoporosis [103].
Sclerostin has been considered a target protein to be neutralized with IgG2 monoclonal antibodies, known as romosozumab, originally AMG785, (Amgen Inc., Thousand Oaks, CA, USA; and UCB Pharma, Brussels, Belgium), which activates Wnt signaling, thereby increasing bone formation, inhibiting bone resorption, and increasing bone mass in patients with osteoporosis [104]. Considerable interest has emerged in this anti-sclerostin antibody ever since the first clinical trial was reported in 2011 by Padhi et al. [105]. Two major phase III trials, by Cosman et al. [106] and Saag et al. [107], demonstrated that treatment with romosozumab decreased the incidence of fractures in patients with osteoporosis. Both studies proposed romosozumab as a promising therapy in patients with osteoporosis [108]. These extensive trials and expanded evidence of the beneficial effects of anti-sclerostin treatment led to its approval in the US, EU, Canada, and Japan in 2019. Treatment with romosozumab has also been suggested to have a positive effect on bone diseases associated with multiple myeloma [109]. Patients with plasma cell malignancies such as multiple myeloma have elevated levels of sclerostin, which may contribute to the pathophysiology of lytic bone lesions associated with this hematological malignancy [110,111].
6. Current Assays—Sclerostin and Bioactive Sclerostin
Although not clinically proven, as of today, the assessment of circulating sclerostin may have a potential role in the routine clinical setting for different disorders affecting bone remodeling and for monitoring patients treated with different therapeutic modalities. A large number of assays are available (for research use) for the measurement of circulating (Table 1). However, there is little knowledge about how results from different assays are concordant with each other, which confounds direct comparisons between different clinical trials applying various sclerostin assays. Piec et al. [112] revealed significant variability in three different sclerostin assays arguing the accuracy and specificity of these assays. Hitherto, only a few assays have been developed to measure the bioactive/intact sclerostin (i.e., Biomedica (Vienna, Austria), DiaSorin Inc. (Stillwater, MN, USA), and TECOmedical (Sissach, Switzerland)).

Table 1.
Commercially available assays for determination of circulating human sclerostin.
Most of the available sclerostin tests are provided as enzyme-linked immunosorbent assays (ELISA) that notably differ regarding assay range, sensitivity, and turnaround time (Table 1). Except for a few companies, most manufacturers do not provide specific information regarding the traceability of the standards used in their assays. As shown in Table 1, two ELISA kits from different manufacturers, i.e., G-Biosciences (St. Louis, MO, USA) and LifeSpan BioSciences (Seattle, WA, USA) have exactly the same assay range, which indicates that they may have identical standards from the same manufacturer but referred to different assay brand names; however, we did not confirm this by contacting the manufacturers. An alternative to ELISA is provided by Meso Scale Discovery (Rockville, MD, USA) implementing multiplex analysis (Human Bone Panel I Kit) in which sclerostin is included besides two other bone biomarkers (i.e., osteoprotegerin and alkaline phosphatase).
To our knowledge, the LIAISON® sclerostin (DiaSorin Inc.) chemiluminescence immunoassay (CLIA) is the only fully automated method available. This assay is designed to detect only intact sclerostin and has the shortest turnaround time (1 h) of all assays. Drake et al. [113] compared the LIAISON® sclerostin assay with two other methods, namely Biomedica sclerostin and Meso Scale Discovery (Human Bone Panel I Kit), which demonstrated that the LIAISON® sclerostin CLIA has an enhanced sensitivity and matched plasma and serum equivalence. It should, however, be noted that Biomedica recently has developed and released an assay for bioactive (intact) sclerostin that might be comparable with the intact sclerostin LIAISON® assay.
Another study by Durosier et al. [114] on 189 healthy adults showed that sclerostin levels were markedly different when measured by three different assays, namely Meso Scale Discovery, Biomedica, and TECOmedical. The Meso Scale Discovery assay resulted in low absolute values, which the authors suggested could be due to this assay specifically detecting intact sclerostin, while the two other assays detect circulating fragments of sclerostin [114,115]. However, the antibody specificity for the Meso Scale Discovery assay towards intact sclerostin has been questioned by others and, importantly, the assay protocol does not state that this method is specific for intact sclerostin [113].
Taken together, the value of assessing sclerostin in the routine clinical setting has yet to be shown and described discrepancies between assays will influence the use of sclerostin in clinical practice. Reported immunoassay variabilities for the measurement of sclerostin increase the need for further comparative studies that may improve the understanding of sclerostin in clinical studies. Although not investigated, it is likely that results from different sclerostin assays cannot be compared with each other even if normalized against healthy individuals. A long-term objective to overcome problems with discrepancies between sclerostin values generated by different assays would be to design projects working on assay harmonization or standardization. The International Osteoporosis Foundation (IOF) and the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) have established a working group, i.e., the IOF-IFCC Joint Working Group on Bone Marker Standards (a.k.a. WG-BMS). This working group has performed significant work towards the harmonization of the bone markers type I procollagen intact N-terminal propeptide (a.k.a. PINP) assays and the standardization of CTX assays in collaboration with the assay manufacturing industry [116], and would thus be the appropriate forum for a project regarding sclerostin assays.
Sclerostin Reference Intervals
Reported reference intervals for serum and plasma sclerostin in adult individuals are presented in Table 2. Besides peer-reviewed articles, Table 2 also includes information about reference intervals given by different companies in the instructions for use. The circulating levels of sclerostin are associated with age and sex where males, in general, have higher sclerostin levels than females across the adult lifespan. These sex-specific differences are also noted during childhood and adolescence, particularly during early puberty where boys have higher sclerostin levels in comparison with girls and, later on, serum sclerostin levels decline in late puberty in both sexes [117]. It is therefore essential to use age- and sex-specific reference intervals for the interpretation of sclerostin in all age groups. Osteogenesis differs in children and adolescents from that of adult individuals since it, besides remodeling, also covers modeling and linear growth of the skeleton. These developmental differences are reflected by biomarkers of bone metabolism and it has been widely reported that children, and in particular adolescents during the pubertal growth spurt, have higher circulating levels of biochemical bone markers of bone turnover in comparison with adults [118]. Grouping biochemical tests by means of Tanner stage partitioning is a complementary and alternative approach to age- and sex-specific reference intervals that can be applied because it reflects bone growth and development that influence biomarkers of bone metabolism [119]. Reflecting the methodological characteristics and reported discrepancies between assays (described above), it is also pivotal to use assay-specific reference intervals when evaluating and comparing measured sclerostin values.
Most sclerostin assays have validated reference intervals for both serum and plasma samples, and it should be observed that measurements of sclerostin are consistently higher in plasma in comparison with serum samples (Table 2). The reason for this observation is currently unknown, but it was initially suggested that this was due to molecular interactions between heparin and reported heparin-binding sites on sclerostin, which potentially could interfere with sclerostin binding proteins by exposing recognizable epitopes for assay antibodies towards sclerostin [56]. However, later studies on EDTA and citrate plasma revealed that blood samples collected with these additives also have higher levels of sclerostin in comparison with serum samples (Table 2) [120].
Table 2.
Reported reference intervals for serum and plasma sclerostin in adult individuals.
Table 2.
Reported reference intervals for serum and plasma sclerostin in adult individuals.
| Reference/Source (Year) | Assays | Reference Interval a (pmol/L) |
|---|---|---|
| Mödder et al. [75] (2011) | Biomedica | Healthy subjects, 21–97 years Pre-menopausal females, n = 123: 24 ± 1 b Post-menopausal females, n = 152: 28 ± 1 Males, n = 318: 33 ± 1 |
| McNulty et al. [121] (2011) | Biomedica | Healthy subjects, 20–59 years, n = 25 Serum: 44 ± 5; heparin plasma: 65 ± 6 b |
| Ardawi et al. [122] (2011) | Biomedica | Pre-menopausal females, n = 1235: 7.5–46 Post-menopausal females, n = 568: 23–74 |
| Durosier et al. [114] (2013) | Biomedica Meso Scale Discovery | Healthy subjects, 65 ± 1 years, n = 187 Biomedica: 20–101 Meso Scale Discovery: 0.79–3.0 |
| Piec et al. [112] (2016) | Biomedica TECOmedical, High sensitive R&D Systems | Healthy subjects, 18–26 years, n = 46 Biomedica: 22–59 (EDTA plasma: 23–71) c TECOmedical: 11–33 (EDTA plasma: 14–49) R&D Systems: 2.7–13 (EDTA plasma: 15–53) |
| Drake et al. [113] (2018) | LIAISON® Sclerostin | Healthy subjects, 21–97 years Females, n = 265: 6.2–36 Males, n = 271: 8.2–51 |
| Kerschan-Schindl et al. [123] (2022) | Biomedica (Bioactive sclerostin) | Healthy subjects, 26–74 years Females, n = 175: 51 (38–70) d Males, n = 61: 62 (41–92) |
| Biomedica (instructions for use) | Sclerostin, BI-20492 | Healthy subjects, n = 411: 11–52 |
| Biomedica (instructions for use) | Bioactive sclerostin BI-20472 | Healthy subjects Serum, n = 32, 12–143 EDTA plasma, n = 24, 29–226 |
| R&D Systems (Instructions for use) | Quantikine, DSST00 | Healthy subjects, n = 35 Serum: 2.9–13 EDTA plasma: 8.8–31 Heparin plasma: 9.8–35 |
| TECOmedical (Instructions for use) | TECO® High sensitive | Pre-menopausal females, n = 20: 9–33 Post-menopausal females, n = 19: 11–40 Males, n = 10: 25–51 |
a Reference intervals given for serum samples if not stated otherwise. b Mean ± SEM. c Minimum–maximum. d Median (interquartile range). Reference intervals presented as the mean ± 2SD, or 95% confidence intervals, if not stated otherwise. All sclerostin values are expressed in pmol/L using the conversion factor of 0.044 from pg/mL to pmol/L, i.e., 1 pg/mL = 0.044 pmol/L. Note that reference intervals for discontinued assays are excluded even if they are reported in the given references.
7. Summary and Conclusions
This narrative review covers the in vitro regulation of human sclerostin and SOST gene expression, structural and functional properties of sclerostin, pathophysiology, biological variability, and recent assay developments for measuring circulating sclerostin. By employing the novel AlphaFold Protein Structure Database, with a deep learning algorithm voted Method-of-the-Year 2021 in Nature Methods [60], we generated a comprehensive, state-of-the-art three-dimensional structure prediction for human sclerostin. Reported methodological discrepancies and inconsistencies among immunoassays for the measurement of sclerostin increase the need for further comparative studies that may improve the understanding of sclerostin in clinical studies. The assessment of circulating sclerostin may have a potential role in the routine clinical setting; however, this has yet to be proven and clinically validated in different disorders affecting bone and mineral metabolism.
Author Contributions
Conceptualization: A.O., D.A. and P.M.; writing—original draft preparation: A.O., D.A., F.L. and P.M.; writing—review and editing: A.O., D.A. and P.M.; visualization: D.A., supervision: P.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by ALF Grants from Region Östergötland.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 1,25(OH)2D | 1,25-dihydroxyvitamin D |
| CLIA | Chemiluminescence immunoassay |
| CTX | C-terminal telopeptide of type I collagen |
| Cx43 | Connexin 43 |
| DKK1 | Dickkopf-1 |
| ECM | Extracellular matrix |
| ECR5 | Evolutionarily conserved region 5 |
| ELISA | Enzyme-linked immunosorbent assay |
| EP2 | Prostaglandin E2 receptor subtype 2 |
| EP4 | Prostaglandin E2 receptor subtype 4 |
| ER | Estrogen receptor |
| ERK | Extracellular signal-regulated kinase |
| FAK | Focal adhesion kinase |
| FFSS | Fluid flow shear stress |
| HDAC4/5 | Histone deacetylase 4/5 |
| LRP | Low-density lipoprotein receptor-related protein |
| MEF2C | Myocyte-specific enhancer factor 2C |
| PDB | Paget’s disease of bone |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphoinositide 3-kinase |
| pLDDT | Predicted local-distance difference test |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| Wnt | Wingless-related integration site |
References
- Van Hul, W.; Balemans, W.; Van Hul, E.; Dikkers, F.G.; Obee, H.; Stokroos, R.J.; Hildering, P.; Vanhoenacker, F.; Van Camp, G.; Willems, P.J. Van Buchem disease (hyperostosis corticalis generalisata) maps to chromosome 17q12-q21. Am. J. Hum. Genet. 1998, 62, 391–399. [Google Scholar] [CrossRef]
- Van Buchem, F.S.; Hadders, H.N.; Ubbens, R. An uncommon familial systemic disease of the skeleton: Hyperostosis corticalis generalisata familiaris. Acta Radiol. 1955, 44, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Truswell, A.S. Osteopetrosis with syndactyly; a morphological variant of Albers-Schönberg’s disease. J. Bone Jt. Surg. Br. Vol. 1958, 40, 209–218. [Google Scholar]
- Beighton, P.; Barnard, A.; Hamersma, H.; van der Wouden, A. The syndromic status of sclerosteosis and van Buchem disease. Clin. Genet. 1984, 25, 175–181. [Google Scholar] [CrossRef]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef]
- Balemans, W.; Van Den Ende, J.; Paes-Alves, A.F.; Dikkers, F.G.; Willems, P.J.; Vanhoenacker, F.; de Almeido-Melo, N.; Alves, C.F.; Stratakis, C.A.; Hill, S.C.; et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am. J. Hum. Genet. 1999, 64, 1661–1669. [Google Scholar] [CrossRef]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef]
- Loots, G.G.; Kneissel, M.; Keller, H.; Baptist, M.; Chang, J.; Collette, N.M.; Ovcharenko, D.; Plajzer-Frick, I.; Rubin, E.M. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005, 15, 928–935. [Google Scholar] [CrossRef]
- van Lierop, A.H.; Appelman-Dijkstra, N.M.; Papapoulos, S.E. Sclerostin deficiency in humans. Bone 2017, 96, 51–62. [Google Scholar] [CrossRef]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Ramos Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef]
- Chang, M.-K.; Kramer, I.; Huber, T.; Kinzel, B.; Guth-Gundel, S.; Leupin, O.; Kneissel, M. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc. Natl. Acad. Sci. USA 2014, 111, E5187–E5195. [Google Scholar] [CrossRef]
- Fijalkowski, I.; Geets, E.; Steenackers, E.; Van Hoof, V.; Ramos, F.J.; Mortier, G.; Fortuna, A.M.; Van Hul, W.; Boudin, E. A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J. Bone Miner. Res. 2016, 31, 874–881. [Google Scholar] [CrossRef]
- Martínez-Gil, N.; Roca-Ayats, N.; Cozar, M.; Garcia-Giralt, N.; Ovejero, D.; Nogués, X.; Grinberg, D.; Balcells, S. Genetics and genomics of SOST: Functional analysis of variants and genomic regulation in osteoblasts. Int. J. Mol. Sci. 2021, 22, 489. [Google Scholar] [CrossRef]
- Ye, W.; Wang, Y.; Mei, B.; Hou, S.; Liu, X.; Wu, G.; Qin, L.; Zhao, K.; Huang, Q. Computational and functional characterization of four SNPs in the SOST locus associated with osteoporosis. Bone 2018, 108, 132–144. [Google Scholar] [CrossRef]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef]
- Buenzli, P.R.; Sims, N.A. Quantifying the osteocyte network in the human skeleton. Bone 2015, 75, 144–150. [Google Scholar] [CrossRef]
- Robling, A.G.; Bonewald, L.F. The osteocyte: New insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Chailakhyan, R.K.; Latsinik, N.V.; Panasyuk, A.F.; Keiliss-Borok, I.V. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues: Cloning in vitro and retransplantation in vivo. Transplantation 1974, 17, 331–340. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Bianco, P.; Robey, P.G. Skeletal stem cells. Development 2015, 142, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.M. The mechanism of coupling: A role for the vasculature. Bone 2000, 26, 319–323. [Google Scholar] [CrossRef]
- Riggs, B.L.; Parfitt, A.M. Drugs used to treat osteoporosis: The critical need for a uniform nomenclature based on their action on bone remodeling. J. Bone Miner. Res. 2005, 20, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Youlten, S.E.; Kemp, J.P.; Logan, J.G.; Ghirardello, E.J.; Sergio, C.M.; Dack, M.R.G.; Guilfoyle, S.E.; Leitch, V.D.; Butterfield, N.C.; Komla-Ebri, D.; et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 2021, 12, 2444. [Google Scholar] [CrossRef]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef]
- van Bezooijen, R.L.; Deruiter, M.C.; Vilain, N.; Monteiro, R.M.; Visser, A.; van der Wee-Pals, L.; van Munsteren, C.J.; Hogendoorn, P.C.; Aguet, M.; Mummery, C.L.; et al. SOST expression is restricted to the great arteries during embryonic and neonatal cardiovascular development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 606–612. [Google Scholar] [CrossRef]
- Collette, N.M.; Yee, C.S.; Murugesh, D.; Sebastian, A.; Taher, L.; Gale, N.W.; Economides, A.N.; Harland, R.M.; Loots, G.G. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev. Biol. 2013, 383, 90–105. [Google Scholar] [CrossRef]
- Sebastian, A.; Loots, G.G. Genetics of Sost/SOST in sclerosteosis and van Buchem disease animal models. Metabolism 2018, 80, 38–47. [Google Scholar] [CrossRef]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef]
- Kim, R.Y.; Yang, H.J.; Song, Y.M.; Kim, I.S.; Hwang, S.J. Estrogen modulates bone morphogenetic protein-induced sclerostin expression through the Wnt signaling pathway. Tissue Eng. Part A 2015, 21, 2076–2088. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.; Lara-Castillo, N.; Akhter, M.P.; Dallas, M.; Scott, J.M.; Ganesh, T.; Johnson, M.L. Osteocyte Wnt/β-catenin pathway activation upon mechanical loading is altered in ovariectomized mice. Bone Rep. 2021, 15, 101129. [Google Scholar] [CrossRef] [PubMed]
- Burger, E.H.; Klein-Nulend, J. Mechanotransduction in bone—Role of the lacuno-canalicular network. FASEB J. 1999, 13, S101–S112. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, J.C.; Senwar, B.; Ferguson, V.L. Spaceflight-induced bone tissue changes that affect bone quality and increase fracture risk. Curr. Osteoporos. Rep. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, T.R.; Siamwala, J.H.; Hargens, A.R.; Macias, B.R. Thirty days of spaceflight does not alter murine calvariae structure despite increased Sost expression. Bone Rep. 2017, 7, 57–62. [Google Scholar] [CrossRef]
- Swolin-Eide, D.; Magnusson, P. Does whole-body vibration treatment make children’s bones stronger? Curr. Osteoporos. Rep. 2020, 18, 471–479. [Google Scholar] [CrossRef]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef]
- Tubic, B.; Zeijlon, R.; Wennergren, G.; Obermayer-Pietsch, B.; Mårild, S.; Dahlgren, J.; Magnusson, P.; Swolin-Eide, D. Randomised study of children with obesity showed that whole body vibration reduced sclerostin. Acta Paediatr. 2019, 108, 502–513. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Bellido, T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat. Rev. Endocrinol. 2016, 12, 593–605. [Google Scholar] [CrossRef]
- Qin, L.; Liu, W.; Cao, H.; Xiao, G. Molecular mechanosensors in osteocytes. Bone Res. 2020, 8, 23. [Google Scholar] [CrossRef]
- Riquelme, M.A.; Gu, S.; Hua, R.; Jiang, J.X. Mechanotransduction via the coordinated actions of integrins, PI3K signaling and Connexin hemichannels. Bone Res. 2021, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.S.; Joca, H.C.; Law, R.A.; Williams, K.M.; Kerr, J.P.; Shi, G.; Khairallah, R.J.; Martin, S.S.; Konstantopoulos, K.; Ward, C.W.; et al. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci. Signal. 2017, 10, eaan5748. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, F.; Hayashi, M.; Mouri, Y.; Nakamura, S.; Adachi, T.; Nakashima, T. Mechanotransduction via the Piezo1-Akt pathway underlies Sost suppression in osteocytes. Biochem. Biophys. Res. Commun. 2020, 521, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Spatz, J.; Nishimori, S.; Doench, J.; Root, D.; Babij, P.; Nagano, K.; Baron, R.; Brooks, D.; Bouxsein, M.; et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J. Bone Miner. Res. 2015, 30, 400–411. [Google Scholar] [CrossRef]
- Sato, T.; Verma, S.; Andrade, C.D.C.; Omeara, M.; Campbell, N.; Wang, J.S.; Cetinbas, M.; Lang, A.; Ausk, B.J.; Brooks, D.J.; et al. A FAK/HDAC5 signaling axis controls osteocyte mechanotransduction. Nat. Commun. 2020, 11, 3282. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.K.; Harris, S.; Ahuja, S.S.; Bonewald, L.F. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res. 2002, 17, 2068–2079. [Google Scholar] [CrossRef]
- Wang, K.; Le, L.; Chun, B.M.; Tiede-Lewis, L.M.; Shiflett, L.A.; Prideaux, M.; Campos, R.S.; Veno, P.A.; Xie, Y.; Dusevich, V.; et al. A novel osteogenic cell line that differentiates into GFP-tagged osteocytes and forms mineral with a bone-like lacunocanalicular structure. J. Bone Miner. Res. 2019, 34, 979–995. [Google Scholar] [CrossRef]
- Uda, Y.; Azab, E.; Sun, N.; Shi, C.; Pajevic, P.D. Osteocyte mechanobiology. Curr. Osteoporos. Rep. 2017, 15, 318–325. [Google Scholar] [CrossRef]
- Lara-Castillo, N.; Kim-Weroha, N.A.; Kamel, M.A.; Javaheri, B.; Ellies, D.L.; Krumlauf, R.E.; Thiagarajan, G.; Johnson, M.L. In vivo mechanical loading rapidly activates β-catenin signaling in osteocytes through a prostaglandin mediated mechanism. Bone 2015, 76, 58–66. [Google Scholar] [CrossRef]
- Williams, K.M.; Leser, J.M.; Gould, N.R.; Joca, H.C.; Lyons, J.S.; Khairallah, R.J.; Ward, C.W.; Stains, J.P. TRPV4 calcium influx controls sclerostin protein loss independent of purinergic calcium oscillations. Bone 2020, 136, 115356. [Google Scholar] [CrossRef]
- Johnson, M.L. Unlocking the sost gene. J. Bone Miner. Res. 2015, 30, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Baertschi, S.; Halleux, C.; Keller, H.; Kneissel, M. Mef2c deletion in osteocytes results in increased bone mass. J. Bone Miner. Res. 2012, 27, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Galea, G.L.; Meakin, L.B.; Sugiyama, T.; Zebda, N.; Sunters, A.; Taipaleenmaki, H.; Stein, G.S.; van Wijnen, A.J.; Lanyon, L.E.; Price, J.S. Estrogen receptor α mediates proliferation of osteoblastic cells stimulated by estrogen and mechanical strain, but their acute down-regulation of the Wnt antagonist Sost is mediated by estrogen receptor β. J. Biol. Chem. 2013, 288, 9035–9048. [Google Scholar] [CrossRef] [PubMed]
- Wijenayaka, A.R.; Yang, D.; Prideaux, M.; Ito, N.; Kogawa, M.; Anderson, P.H.; Morris, H.A.; Solomon, L.B.; Loots, G.G.; Findlay, D.M.; et al. 1α,25-dihydroxyvitamin D3 stimulates human SOST gene expression and sclerostin secretion. Mol. Cell. Endocrinol. 2015, 413, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Wijenayaka, A.R.; Prideaux, M.; Yang, D.; Morris, H.A.; Findlay, D.M.; Anderson, P.H.; Atkins, G.J. Early response of the human SOST gene to stimulation by 1α,25-dihydroxyvitamin D(3). J. Steroid Biochem. Mol. Biol. 2016, 164, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Veverka, V.; Henry, A.J.; Slocombe, P.M.; Ventom, A.; Mulloy, B.; Muskett, F.W.; Muzylak, M.; Greenslade, K.; Moore, A.; Zhang, L.; et al. Characterization of the structural features and interactions of sclerostin. J. Biol. Chem. 2009, 284, 10890–10900. [Google Scholar] [CrossRef]
- Hernandez, P.; Whitty, C.; John Wardale, R.; Henson, F.M. New insights into the location and form of sclerostin. Biochem. Biophys. Res. Commun. 2014, 446, 1108–1113. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Marx, V. Method of the year: Protein structure prediction. Nat. Methods 2022, 19, 5–10. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, J.; Liu, X.; Linhardt, R.J. Interactions between sclerostin and glycosaminoglycans. Glycoconj. J. 2020, 37, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Boschert, V.; van Dinther, M.; Weidauer, S.; van Pee, K.; Muth, E.M.; ten Dijke, P.; Mueller, T.D. Mutational analysis of sclerostin shows importance of the flexible loop and the cystine-knot for Wnt-signaling inhibition. PLoS ONE 2013, 8, e81710. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Han, W.; Park, T.; Kim, E.J.; Bang, I.; Lee, H.S.; Jeong, Y.; Roh, K.; Kim, J.; Kim, J.S.; et al. Sclerostin inhibits Wnt signaling through tandem interaction with two LRP6 ectodomains. Nat. Commun. 2020, 11, 5357. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–w437. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [PubMed]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Westendorf, J.J.; Kahler, R.A.; Schroeder, T.M. Wnt signaling in osteoblasts and bone diseases. Gene 2004, 341, 19–39. [Google Scholar] [CrossRef]
- Glass, D.A., II; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef]
- Hu, H.; Hilton, M.J.; Tu, X.; Yu, K.; Ornitz, D.M.; Long, F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 2005, 132, 49–60. [Google Scholar] [CrossRef]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Wang, S.Y.; Li, Y.N.; Bian, W.J.; Zhang, L.Q.; Li, Y.H.; Long, L.; Liu, X.; Zhang, X.W.; Li, Z.G. Activity of fibroblast-like synoviocytes in rheumatoid arthritis was impaired by dickkopf-1 targeting siRNA. Chin. Med. J. 2020, 133, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, R.E.; Kämpe, A.; Costantini, A.; Alm, J.J.; Magnusson, P.; Mäkitie, O. Biomarkers in WNT1 and PLS3 osteoporosis: Altered concentrations of DKK1 and FGF23. J. Bone Miner. Res. 2020, 35, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Mödder, U.I.; Hoey, K.A.; Amin, S.; McCready, L.K.; Achenbach, S.J.; Riggs, B.L.; Melton, L.J., III; Khosla, S. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J. Bone Miner. Res. 2011, 26, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, A.L.; Zhang, Y.; Yu, S.; Kammerer, C.M.; Nestlerode, C.S.; Chu, Y.; Bunker, C.H.; Patrick, A.L.; Wheeler, V.W.; Miljkovic, I.; et al. Relative influence of heritability, environment and genetics on serum sclerostin. Osteoporos Int. 2014, 25, 905–912. [Google Scholar] [CrossRef]
- Jorde, R.; Stunes, A.K.; Kubiak, J.; Grimnes, G.; Thorsby, P.M.; Syversen, U. Smoking and other determinants of bone turnover. PLoS ONE 2019, 14, e0225539. [Google Scholar] [CrossRef]
- Swanson, C.; Shea, S.A.; Wolfe, P.; Markwardt, S.; Cain, S.W.; Munch, M.; Czeisler, C.A.; Orwoll, E.S.; Buxton, O.M. 24-hour profile of serum sclerostin and its association with bone biomarkers in men. Osteoporos Int. 2017, 28, 3205–3213. [Google Scholar] [CrossRef]
- van der Spoel, E.; Oei, N.; Cachucho, R.; Roelfsema, F.; Berbée, J.F.P.; Blauw, G.J.; Pijl, H.; Appelman-Dijkstra, N.M.; van Heemst, D. The 24-hour serum profiles of bone markers in healthy older men and women. Bone 2019, 120, 61–69. [Google Scholar] [CrossRef]
- Ralston, S.H.; Corral-Gudino, L.; Cooper, C.; Francis, R.M.; Fraser, W.D.; Gennari, L.; Guañabens, N.; Javaid, M.K.; Layfield, R.; O’Neill, T.W.; et al. Diagnosis and management of Paget’s disease of bone in adults: A clinical guideline. J. Bone Miner. Res. 2019, 34, 579–604. [Google Scholar] [CrossRef]
- Idolazzi, L.; Fassio, A.; Tripi, G.; Braga, V.; Viapiana, O.; Adami, G.; Rossini, M.; Gatti, D. Circulating Dickkopf-1 and sclerostin in patients with Paget’s disease of bone. Clin. Rheumatol. 2017, 36, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Yavropoulou, M.P.; van Lierop, A.H.; Hamdy, N.A.; Rizzoli, R.; Papapoulos, S.E. Serum sclerostin levels in Paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone 2012, 51, 153–157. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fuentes-Calvo, I.; Usategui-Martín, R.; Calero-Paniagua, I.; Moledo-Pouso, C.; García-Ortiz, L.; Pino-Montes, J.D.; González-Sarmiento, R.; Martínez-Salgado, C. Influence of angiogenic mediators and bone remodelling in Paget’s disease of bone. Int. J. Med. Sci. 2018, 15, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; Graciolli, F.G.; O’Brien, S.; Tang, W.; dos Reis, L.M.; Ryan, S.; Phillips, L.; Boulanger, J.; Song, W.; Bracken, C.; et al. Repression of osteocyte Wnt/β-catenin signaling is an early event in the progression of renal osteodystrophy. J. Bone Miner. Res. 2012, 27, 1757–1772. [Google Scholar] [CrossRef]
- Catalano, A.; Bellone, F.; Morabito, N.; Corica, F. Sclerostin and vascular pathophysiology. Int. J. Mol. Sci. 2020, 21, 4779. [Google Scholar] [CrossRef]
- Drüeke, T.B.; Massy, Z.A. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 2016, 89, 289–302. [Google Scholar] [CrossRef]
- Morena, M.; Jaussent, I.; Dupuy, A.M.; Bargnoux, A.S.; Kuster, N.; Chenine, L.; Leray-Moragues, H.; Klouche, K.; Vernhet, H.; Canaud, B.; et al. Osteoprotegerin and sclerostin in chronic kidney disease prior to dialysis: Potential partners in vascular calcifications. Nephrol. Dial. Transplant. 2015, 30, 1345–1356. [Google Scholar] [CrossRef]
- Cejka, D.; Jäger-Lansky, A.; Kieweg, H.; Weber, M.; Bieglmayer, C.; Haider, D.G.; Diarra, D.; Patsch, J.M.; Kainberger, F.; Bohle, B.; et al. Sclerostin serum levels correlate positively with bone mineral density and microarchitecture in haemodialysis patients. Nephrol. Dial. Transplant. 2012, 27, 226–230. [Google Scholar] [CrossRef]
- Pelletier, S.; Dubourg, L.; Carlier, M.C.; Hadj-Aissa, A.; Fouque, D. The relation between renal function and serum sclerostin in adult patients with CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 819–823. [Google Scholar] [CrossRef]
- Boltenstål, H.; Qureshi, A.R.; Behets, G.J.; Lindholm, B.; Stenvinkel, P.; D’Haese, P.C.; Haarhaus, M. Association of serum sclerostin with bone sclerostin in chronic kidney disease is lost in glucocorticoid treated patients. Calcif. Tissue Int. 2019, 104, 214–223. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Komaba, H.; Hamano, N.; Tanaka, H.; Wada, T.; Ishida, H.; Nakamura, M.; Takahashi, H.; Takahashi, Y.; Hyodo, T.; et al. Interrelationships between sclerostin, secondary hyperparathyroidism, and bone metabolism in patients on hemodialysis. J. Clin. Endocrinol. Metab. 2022, 107, e95–e105. [Google Scholar] [CrossRef] [PubMed]
- Napoli, N.; Incalzi, R.A.; De Gennaro, G.; Marcocci, C.; Marfella, R.; Papalia, R.; Purrello, F.; Ruggiero, C.; Tarantino, U.; Tramontana, F.; et al. Bone fragility in patients with diabetes mellitus: A consensus statement from the working group of the Italian Diabetes Society (SID), Italian Society of Endocrinology (SIE), Italian Society of Gerontology and Geriatrics (SIGG), Italian Society of Orthopaedics and Traumatology (SIOT). Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1375–1390. [Google Scholar] [CrossRef] [PubMed]
- Fuusager, G.; Milandt, N.; Shanbhogue, V.V.; Hermann, A.P.; Schou, A.J.; Christesen, H.T. Lower estimated bone strength and impaired bone microarchitecture in children with type 1 diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001384. [Google Scholar] [CrossRef] [PubMed]
- Novak, D.; Forsander, G.; Kristiansen, E.; Svedlund, A.; Magnusson, P.; Swolin-Eide, D. Altered cortical bone strength and lean mass in young women with long-duration (19 years) type 1 diabetes. Sci. Rep. 2020, 10, 22367. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, C.; Colangelo, L.; Santori, R.; Renella, M.; Mastrantonio, M.; Minisola, S.; Pepe, J. The interplay between bone and glucose metabolism. Front. Endocrinol. 2020, 11, 122. [Google Scholar] [CrossRef]
- Lin, X.; Onda, D.A.; Yang, C.H.; Lewis, J.R.; Levinger, I.; Loh, K. Roles of bone-derived hormones in type 2 diabetes and cardiovascular pathophysiology. Mol. Metab. 2020, 40, 101040. [Google Scholar] [CrossRef]
- Murray, C.E.; Coleman, C.M. Impact of diabetes mellitus on bone health. Int. J. Mol. Sci. 2019, 20, 4873. [Google Scholar] [CrossRef]
- Wędrychowicz, A.; Sztefko, K.; Starzyk, J.B. Sclerostin and its significance for children and adolescents with type 1 diabetes mellitus (T1D). Bone 2019, 120, 387–392. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamauchi, M.; Sugimoto, T. Elevated sclerostin levels are associated with vertebral fractures in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2013, 98, 4030–4037. [Google Scholar] [CrossRef]
- Gaudio, A.; Privitera, F.; Battaglia, K.; Torrisi, V.; Sidoti, M.H.; Pulvirenti, I.; Canzonieri, E.; Tringali, G.; Fiore, C.E. Sclerostin levels associated with inhibition of the Wnt/β-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 3744–3750. [Google Scholar] [CrossRef]
- García-Martín, A.; Rozas-Moreno, P.; Reyes-García, R.; Morales-Santana, S.; García-Fontana, B.; García-Salcedo, J.A.; Muñoz-Torres, M. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2012, 97, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Kanis, J.A.; Delmas, P.; Burckhardt, P.; Cooper, C.; Torgerson, D. Guidelines for diagnosis and management of osteoporosis. The European Foundation for Osteoporosis and Bone Disease. Osteoporos Int. 1997, 7, 390–406. [Google Scholar] [CrossRef] [PubMed]
- Eastell, R.; Szulc, P. Use of bone turnover markers in postmenopausal osteoporosis. Lancet Diabetes Endocrinol. 2017, 5, 908–923. [Google Scholar] [CrossRef]
- Tanaka, S.; Matsumoto, T. Sclerostin: From bench to bedside. J. Bone Miner. Metab. 2021, 39, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Padhi, D.; Jang, G.; Stouch, B.; Fang, L.; Posvar, E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J. Bone Miner. Res. 2011, 26, 19–26. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab treatment in postmenopausal women with osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef]
- Khosla, S. Bone diseases: Romosozumab—on track or derailed? Nat. Rev. Endocrinol. 2017, 13, 697–698. [Google Scholar] [CrossRef]
- Toscani, D.; Bolzoni, M.; Ferretti, M.; Palumbo, C.; Giuliani, N. Role of osteocytes in myeloma bone disease: Anti-sclerostin antibody as new therapeutic strategy. Front. Immunol. 2018, 9, 2467. [Google Scholar] [CrossRef]
- Mabille, C.; Ruyssen-Witrand, A.; Degboe, Y.; Gennero, I.; Loiseau, H.A.; Roussel, M.; Hebraud, B.; Nigon, D.; Attal, M.; Laroche, M. DKK1 and sclerostin are early markers of relapse in multiple myeloma. Bone 2018, 113, 114–117. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Bellido, T.; Roodman, G.D. Role of osteocytes in multiple myeloma bone disease. Curr. Opin. Support. Palliat. Care 2014, 8, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Piec, I.; Washbourne, C.; Tang, J.; Fisher, E.; Greeves, J.; Jackson, S.; Fraser, W.D. How accurate is your sclerostin measurement? Comparison between three commercially available sclerostin ELISA kits. Calcif. Tissue Int. 2016, 98, 546–555. [Google Scholar] [CrossRef]
- Drake, M.T.; Fenske, J.S.; Blocki, F.A.; Zierold, C.; Appelman-Dijkstra, N.; Papapoulos, S.; Khosla, S. Validation of a novel, rapid, high precision sclerostin assay not confounded by sclerostin fragments. Bone 2018, 111, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Durosier, C.; van Lierop, A.; Ferrari, S.; Chevalley, T.; Papapoulos, S.; Rizzoli, R. Association of circulating sclerostin with bone mineral mass, microstructure, and turnover biochemical markers in healthy elderly men and women. J. Clin. Endocrinol. Metab. 2013, 98, 3873–3883. [Google Scholar] [CrossRef] [PubMed]
- van Lierop, A.H.; Hamdy, N.A.; Hamersma, H.; van Bezooijen, R.L.; Power, J.; Loveridge, N.; Papapoulos, S.E. Patients with sclerosteosis and disease carriers: Human models of the effect of sclerostin on bone turnover. J. Bone Miner. Res. 2011, 26, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Bhattoa, H.P.; Cavalier, E.; Eastell, R.; Heijboer, A.C.; Jørgensen, N.R.; Makris, K.; Ulmer, C.Z.; Kanis, J.A.; Cooper, C.; Silverman, S.L.; et al. Analytical considerations and plans to standardize or harmonize assays for the reference bone turnover markers PINP and β-CTX in blood. Clin. Chim. Acta 2021, 515, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Kirmani, S.; Amin, S.; McCready, L.K.; Atkinson, E.J.; Melton, L.J., III; Müller, R.; Khosla, S. Sclerostin levels during growth in children. Osteoporos Int. 2012, 23, 1123–1130. [Google Scholar] [CrossRef]
- Adeli, K.; Higgins, V.; Trajcevski, K.; White-Al Habeeb, N. The Canadian laboratory initiative on pediatric reference intervals: A CALIPER white paper. Crit. Rev. Clin. Lab. Sci. 2017, 54, 358–413. [Google Scholar] [CrossRef]
- Huang, Y.; Eapen, E.; Steele, S.; Grey, V. Establishment of reference intervals for bone markers in children and adolescents. Clin. Biochem. 2011, 44, 771–778. [Google Scholar] [CrossRef]
- Mause, S.F.; Deck, A.; Hennies, M.; Kaesler, N.; Evenepoel, P.; Boisvert, W.A.; Janssen, U.; Brandenburg, V.M. Validation of commercially available ELISAs for the detection of circulating sclerostin in hemodialysis patients. Discoveries 2016, 4, e55. [Google Scholar] [CrossRef]
- McNulty, M.; Singh, R.J.; Li, X.; Bergstralh, E.J.; Kumar, R. Determination of serum and plasma sclerostin concentrations by enzyme-linked immunoassays. J. Clin. Endocrinol. Metab. 2011, 96, E1159–E1162. [Google Scholar] [CrossRef] [PubMed]
- Ardawi, M.S.; Al-Kadi, H.A.; Rouzi, A.A.; Qari, M.H. Determinants of serum sclerostin in healthy pre- and postmenopausal women. J. Bone Miner. Res. 2011, 26, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Kerschan-Schindl, K.; Föger-Samwald, U.; Gleiss, A.; Kudlacek, S.; Wallwitz, J.; Pietschmann, P. Circulating bioactive sclerostin levels in an Austrian population-based cohort. Wien. Klin. Wochenschr. 2022, 134, 39–44. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).