
Novel Chemicals Derived from Tadalafil Exhibit PRMT5 Inhibition and Promising Activities against Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Drug Design of Chemicals A, B, and C
2.2. Compounds A, B, and C Bind to the PRMT5 Protein through Different Binding Modes and Inhibit Histone Arginine Methylation
2.3. Compounds A, B, and C Inhibit Breast Cancer Cell Proliferation
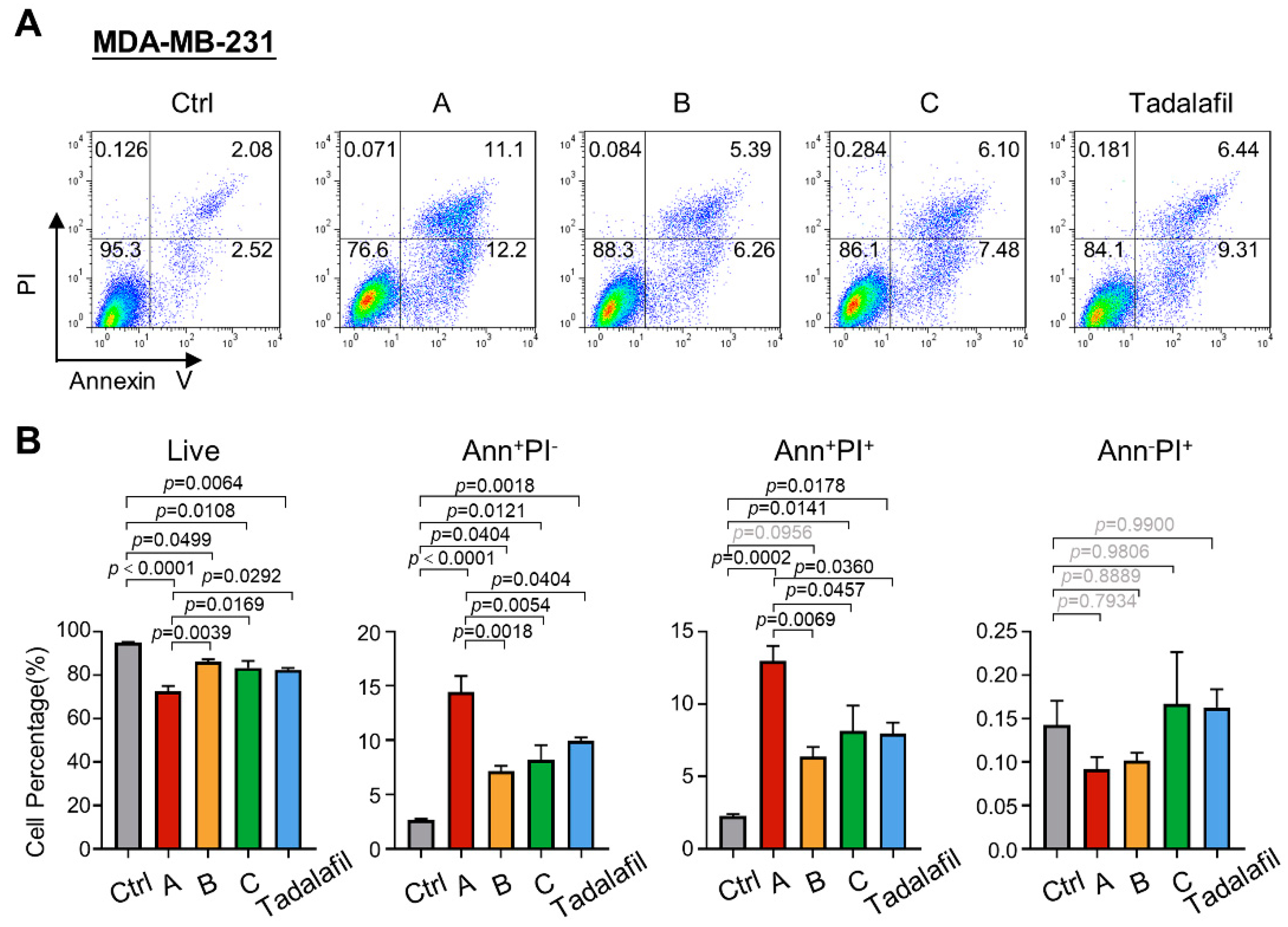
2.4. Compounds A, B, and C Promote Breast Cancer Cell Apoptosis
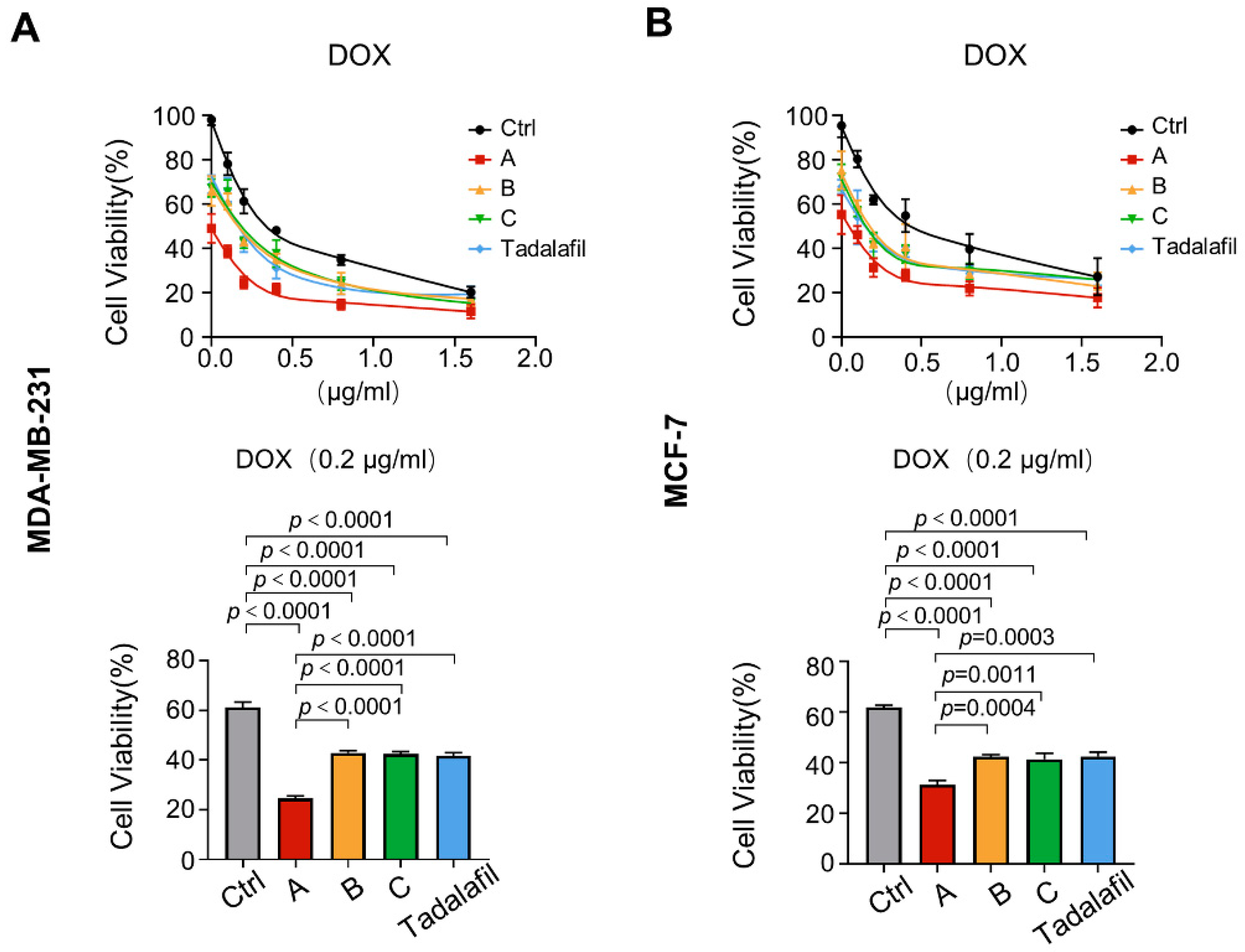
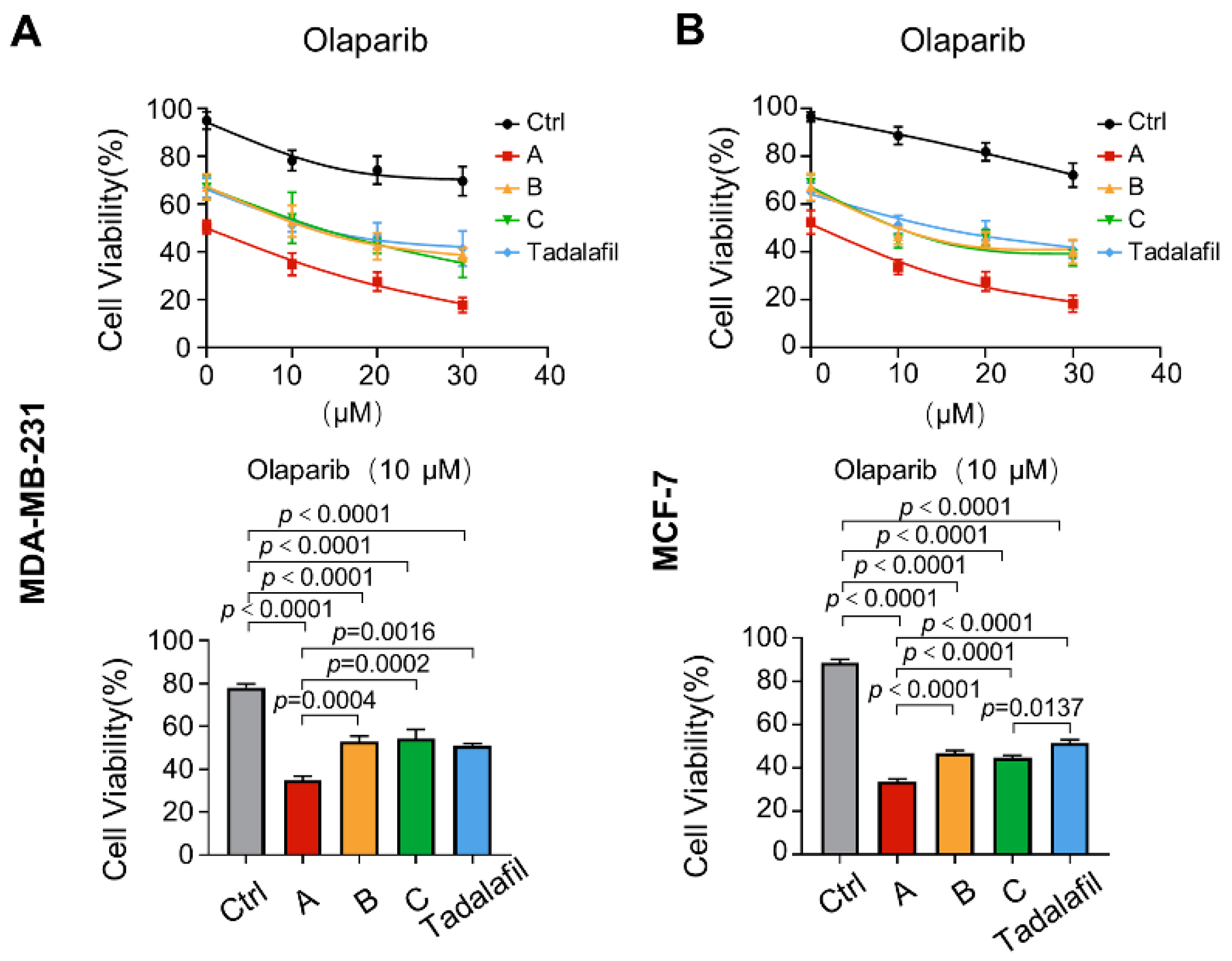
2.5. Compounds A, B, and C Promote the Antitumor Effect of Chemotherapeutics In Vitro
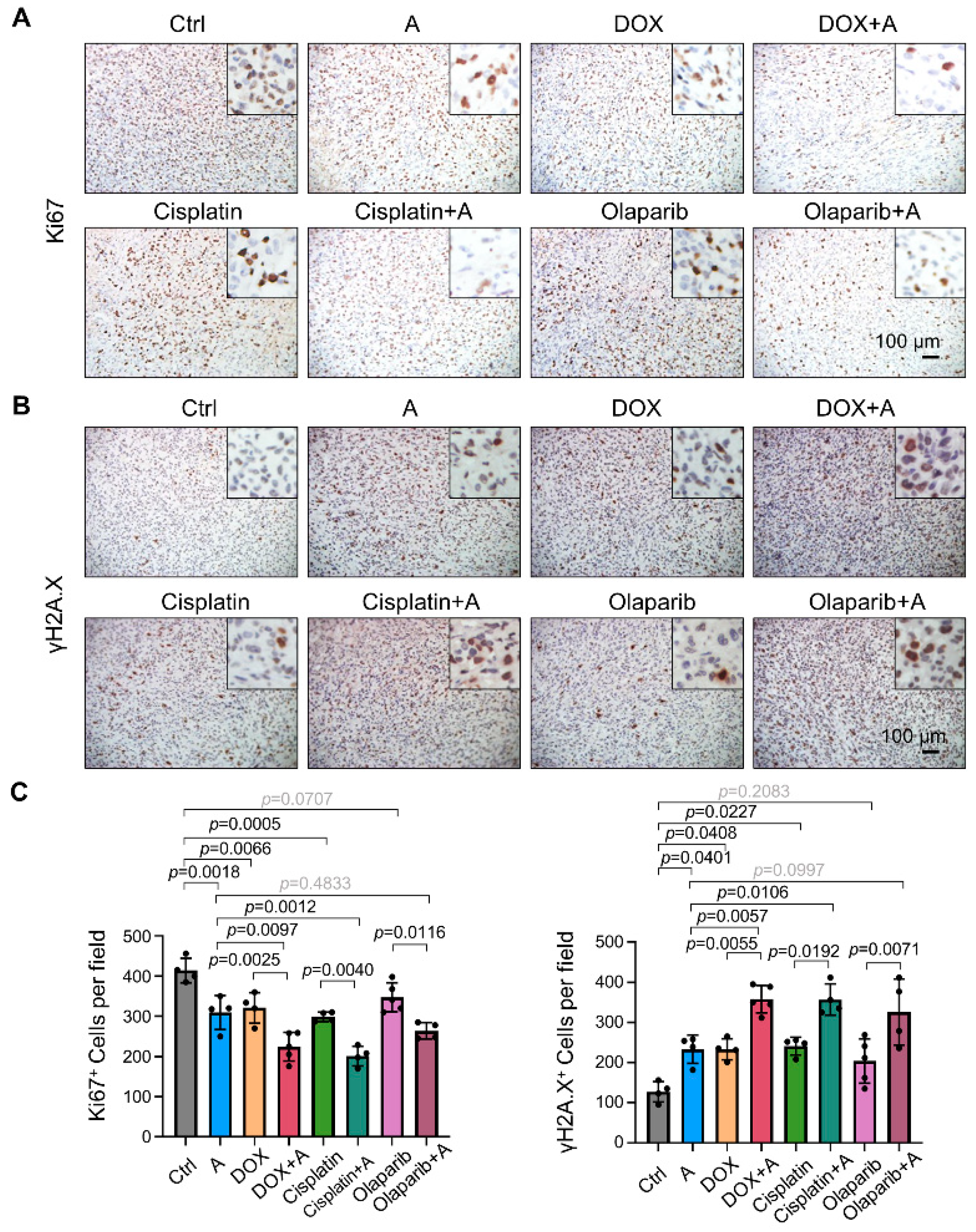
2.6. Compounds A, B, and C Promote the Antitumor Effects of Chemotherapeutics in Xenograft Tumor Models
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.2. SPR Analysis
4.3. Cell Culture
4.4. Cell Proliferation Assays
4.5. Apoptosis Analysis
4.6. Western Blotting
4.7. Mouse Xenograft Model
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Palmieri, C.; Krell, J.; James, C.R.; Harper-Wynne, C.; Misra, V.; Cleator, S.; Miles, D. Rechallenging with anthracyclines and taxanes in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2010, 7, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2021, 27, 436–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, J.; Shen, B.; Wei, G. Integrated Chromatin Accessibility and Transcriptome Landscapes of Doxorubicin-Resistant Breast Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 708066. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Yuan, Y.; Nie, H. Protein arginine methyltransferase 5: A potential cancer therapeutic target. Cell. Oncol. 2021, 44, 33–44. [Google Scholar] [CrossRef]
- Burgos, E.; Wilczek, C.; Onikubo, T.; Bonanno, J.B.; Jansong, J.; Reimer, U.; Shechter, D. Histone H2A and H4 N-terminal Tails Are Positioned by the MEP50 WD Repeat Protein for Efficient Methylation by the PRMT5 Arginine Methyltransferase. J. Biol. Chem. 2015, 290, 9674–9689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonysamy, S. The Structure and Function of the PRMT5:MEP50 Complex. Subcell. Biochem. 2017, 83, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shao, X.; Zhao, X.; Ji, Y.; Liu, X.; Li, P.; Zhang, M.; Wang, Q. Targeting protein arginine methyltransferase 5 in cancers: Roles, inhibitors and mechanisms. Biomed. Pharmacother. 2021, 144, 112252. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, K.M.; Blomquist, C.; Acharya, N.; Li, R.; Ranaghan, M.J.; O’Keefe, M.; Rodriguez, D.J.; Young, M.J.; Kesar, D.; Pal, D.; et al. Molecular basis for substrate recruitment to the PRMT5 methylosome. Mol. Cell 2021, 81, 3481–3495.e7. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chitnis, N.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; Yang, Y.; Li, Z.; Wasik, M.; Klein-Szanto, A.J.P.; Rustgi, A.K.; et al. PRMT5 Is Required for Lymphomagenesis Triggered by Multiple Oncogenic Drivers. Cancer Discov. 2015, 5, 288–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114.e25. [Google Scholar] [CrossRef] [PubMed]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene-Regulatory Networks Leading to Therapeutic Efficacy in JAK2V617F-Mutant MPN. Cancer Discov. 2020, 10, 1742–1757. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, T.; Wu, Y.; Yang, C.; Li, Y.; Du, G.; He, Y.; Liu, W.; Liu, R.; Chen, C.-H.; et al. Arginine methyltransferase PRMT5 methylates and stabilizes KLF5 via decreasing its phosphorylation and ubiquitination to promote basal-like breast cancer. Cell Death Differ. 2021, 28, 2931–2945. [Google Scholar] [CrossRef] [PubMed]
- Bajbouj, K.; Ramakrishnan, R.; Saber-Ayad, M.; Omar, H.; Sharif-Askari, N.S.; Shafarin, J.; Elmoselhi, A.; Ihmaid, A.; Ali, S.A.; Alalool, A.; et al. PRMT5 Selective Inhibitor Enhances Therapeutic Efficacy of Cisplatin in Lung Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6131. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhao, J.; Wang, J.; Wu, J. PRMT5 promotes colorectal cancer growth by interaction with MCM7. J. Cell. Mol. Med. 2021, 25, 3537–3547. [Google Scholar] [CrossRef]
- Vinet, M.; Suresh, S.; Maire, V.; Monchecourt, C.; Némati, F.; Lesage, L.; Pierre, F.; Ye, M.; Lescure, A.; Brisson, A.; et al. Protein arginine methyltransferase 5: A novel therapeutic target for triple-negative breast cancers. Cancer Med. 2019, 8, 2414–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoumanne, A.; Zhang, J.; Chen, X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009, 37, 4965–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamard, P.-J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.E.; Olsen, S.K.; Ostrowski, M.C.; Gan, W. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 2021, 12, 3444. [Google Scholar] [CrossRef]
- Shailesh, H.; Siveen, K.S.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) activates WNT/β-catenin signalling in breast cancer cells via epigenetic silencing of DKK1 and DKK3. J. Cell. Mol. Med. 2021, 25, 1583–1600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kong, J.; Wu, Y.; Zhang, J.; Wang, T.; Li, N.; Fan, J.; Wang, H.; Zhang, J.; Ling, R. PRMT5 determines the sensitivity to chemotherapeutics by governing stemness in breast cancer. Breast Cancer Res. Treat. 2017, 168, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Z.; Han, L.; Guo, Z.; Yan, B.; Guo, L.; Zhao, H.; Wei, M.; Hou, N.; Ye, J.; et al. PRMT5 regulates RNA m6A demethylation for doxorubicin sensitivity in breast cancer. Mol. Ther. 2022; in Press. [Google Scholar] [CrossRef]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Palte, R.L.; Schneider, S.E.; Altman, M.D.; Hayes, R.P.; Kawamura, S.; Lacey, B.M.; Mansueto, M.S.; Reutershan, M.; Siliphaivanh, P.; Sondey, C.; et al. Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5). ACS Med. Chem. Lett. 2020, 11, 1688–1693. [Google Scholar] [CrossRef]
- Jensen-Pergakes, K.; Tatlock, J.; Maegley, K.A.; McAlpine, I.J.; McTigue, M.A.; Xie, T.; Dillon, C.P.; Wang, Y.; Yamazaki, S.; Spiegel, N.; et al. SAM-Competitive PRMT5 Inhibitor PF-06939999 Demonstrates Antitumor Activity in Splicing Dysregulated NSCLC with Decreased Liability of Drug Resistance. Mol. Cancer Ther. 2021, 21, 3–15. [Google Scholar] [CrossRef]
- Arif, S.A.; Poon, H. Tadalafil: A Long-Acting Phosphodiesterase-5 Inhibitor for the Treatment of Pulmonary Arterial Hypertension. Clin. Ther. 2011, 33, 993–1004. [Google Scholar] [CrossRef]
- Peak, T.; Richman, A.; Gur, S.; Yafi, F.A.; Hellstrom, W.J. The Role of PDE5 Inhibitors and the NO/cGMP Pathway in Cancer. Sex. Med. Rev. 2016, 4, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Escribano Subías, P.; Aurtenetxe Pérez, A.; Pérez Olivares, C.; Gómez Climent, L.; Diago Cabezudo, J.I.; Perelló, M.F. Recent advances in the management of pulmonary arterial hypertension: Lessons from the upfront combination of ambrisentan and tadalafil. Expert Rev. Respir. Med. 2021, 15, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Struck, A.-W.; Thompson, M.L.; Wong, L.S.; Micklefield, J. ChemInform Abstract: S-Adenosyl-Methionine-Dependent Methyltransferases: Highly Versatile Enzymes in Biocatalysis, Biosynthesis and Other Biotechnological Applications. Chembiochem 2013, 44, 2642–2655. [Google Scholar] [CrossRef]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hu, W.; Yuan, Y. Protein Arginine Methyltransferase 5 (PRMT5) as an Anticancer Target and Its Inhibitor Discovery. J. Med. Chem. 2018, 61, 9429–9441. [Google Scholar] [CrossRef]
- Zhu, K.; Jiang, C.-S.; Hu, J.; Liu, X.; Yan, X.; Tao, H.; Luo, C.; Zhang, H. Interaction assessments of the first S-adenosylmethionine competitive inhibitor and the essential interacting partner methylosome protein 50 with protein arginine methyltransferase 5 by combined computational methods. Biochem. Biophys. Res. Commun. 2018, 495, 721–727. [Google Scholar] [CrossRef]
- Pal, S.; Baiocchi, R.A.; Byrd, J.C.; Grever, M.R.; Jacob, S.T.; Sif, S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yang, J.; Chen, C.; Li, Z.; Chen, Y.; Zhang, X.; Wang, L.; Zhou, J. Polyphyllin III-Induced Ferroptosis in MDA-MB-231 Triple-Negative Breast Cancer Cells can Be Protected Against by KLF4-Mediated Upregulation of xCT. Front. Pharmacol. 2021, 12, 670224. [Google Scholar] [CrossRef] [PubMed]
- Rachakhom, W.; Khaw-On, P.; Pompimon, W.; Banjerdpongchai, R. Dihydrochalcone Derivative Induces Breast Cancer Cell Apoptosis via Intrinsic, Extrinsic, and ER Stress Pathways but Abolishes EGFR/MAPK Pathway. BioMed Res. Int. 2019, 2019, 7298539. [Google Scholar] [CrossRef] [PubMed]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.S.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Luo, Q.; Zhang, Y.; Jia, F.; Zhao, Y.; Wang, F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem. Res. Toxicol. 2019, 32, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Abadi, A.J.; Mirzaei, S.; Mahabady, M.K.; Hashemi, F.; Zabolian, A.; Hashemi, F.; Raee, P.; Aghamiri, S.; Ashrafizadeh, M.; Aref, A.R.; et al. Curcumin and its derivatives in cancer therapy: Potentiating antitumor activity of cisplatin and reducing side effects. Phytotherapy Res. 2021, 36, 189–213. [Google Scholar] [CrossRef]
- Montoni, A.; Robu, M.; Pouliot, E.; Shah, G.M. Resistance to PARP-Inhibitors in Cancer Therapy. Front. Pharmacol. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricks, T.K.; Chiu, H.-J.; Ison, G.; Kim, G.; McKee, A.E.; Kluetz, P.; Pazdur, R. Successes and Challenges of PARP Inhibitors in Cancer Therapy. Front. Oncol. 2015, 5, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Wang, Y.; Huang, H.; Li, W.; Ma, J.; Yao, D.; Tang, Z.; Xue, T.; Ha, L.; Ren, Y.; et al. The Tetramethylpyrazine Derivative Statmp-151: A Novel Small Molecule Stat3 Inhibitor with Promising Activity against Breast Cancer. Front. Pharmacol. 2021, 12, 651976. [Google Scholar] [CrossRef]
- Griesbach, E.; Schlackow, M.; Marzluff, W.F.; Proudfoot, N.J. Dual RNA 3’-end processing of H2A.X messenger RNA maintains DNA damage repair throughout the cell cycle. Nat. Commun. 2021, 12, 359. [Google Scholar] [CrossRef]
- Marquette, C.; Nabell, L. Chemotherapy-Resistant Metastatic Breast Cancer. Curr. Treat. Options Oncol. 2012, 13, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Jamialahmadi, K.; Zahedipour, F.; Karimi, G. The role of microRNAs on doxorubicin drug resistance in breast cancer. J. Pharm. Pharmacol. 2021, 73, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wick, N.; Germans, S.K.; Peng, Y. The Role of Breast Cancer Stem Cells in Chemoresistance and Metastasis in Triple-Negative Breast Cancer. Cancers 2021, 13, 6209. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Scatena, C.; Ghilli, M.; Bargagna, I.; Lorenzini, G.; Nicolini, A. Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. Int. J. Mol. Sci. 2022, 23, 1665. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Barchiesi, G.; Roberto, M.; Verrico, M.; Vici, P.; Tomao, S.; Tomao, F. Emerging Role of PARP Inhibitors in Metastatic Triple Negative Breast Cancer. Current Scenario and Future Perspectives. Front. Oncol. 2021, 11, 769280. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.-W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Xiao, T.; Li, Z.; Zhang, J.; Chen, S. Novel Chemicals Derived from Tadalafil Exhibit PRMT5 Inhibition and Promising Activities against Breast Cancer. Int. J. Mol. Sci. 2022, 23, 4806. https://doi.org/10.3390/ijms23094806
Yang Z, Xiao T, Li Z, Zhang J, Chen S. Novel Chemicals Derived from Tadalafil Exhibit PRMT5 Inhibition and Promising Activities against Breast Cancer. International Journal of Molecular Sciences. 2022; 23(9):4806. https://doi.org/10.3390/ijms23094806
Chicago/Turabian StyleYang, Ziyan, Tian Xiao, Zezhi Li, Jian Zhang, and Suning Chen. 2022. "Novel Chemicals Derived from Tadalafil Exhibit PRMT5 Inhibition and Promising Activities against Breast Cancer" International Journal of Molecular Sciences 23, no. 9: 4806. https://doi.org/10.3390/ijms23094806