
Dendritic Cells and Their Immunotherapeutic Potential for Treating Type 1 Diabetes

 and
and
Abstract
:1. Introduction
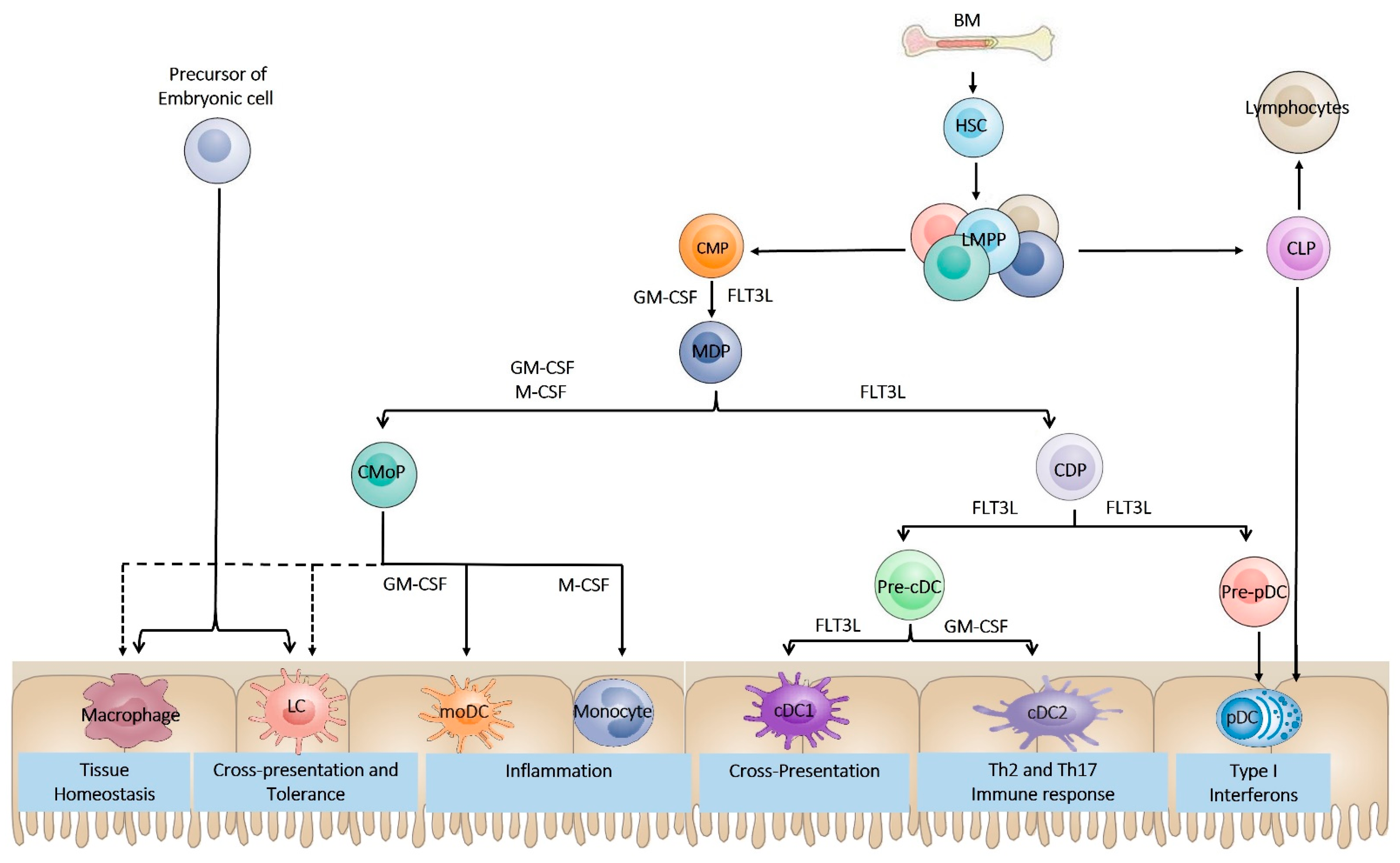
2. Dendritic Cell Ontogeny
3. Dendritic Cell Subsets
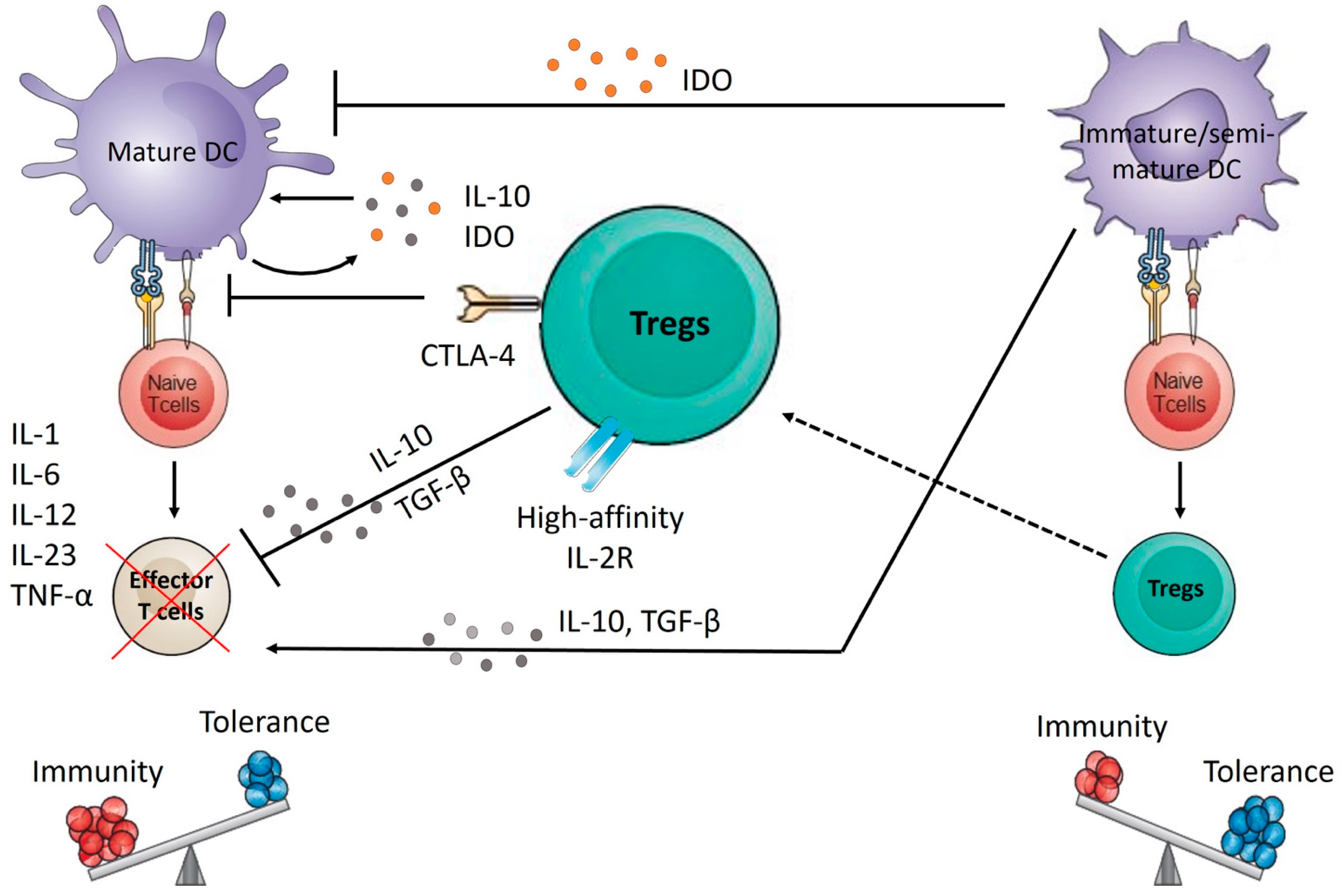
4. Dendritic Cell Plasticity
5. Metabolic Changes in DC during Development, Rest, and Activation
6. Type 1 Diabetes and Dendritic Cells
7. Dendritic Cell-Targeted Therapies for Treating T1D
7.1. Costimulation Blockade
7.2. Blocking Cytokine Production
7.3. In Vivo Targeting of DCs
7.4. Ex Vivo Generation of Tolerogenic DC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Number | Recruitment Status | Study Date | Completion Date | Groups | Outcomes | Reference |
|---|---|---|---|---|---|---|
| NCT00505375 | Completed | February 2008 | May 2012 | Interventional | At the end of the treatment, patients receiving abatacept showed significant C-peptide preservation compared with the placebo group (59% higher, p = 0.0029) at 24 months. However, after 6 months, C-peptide preservation declined to the placebo level, despite continuous treatment for 2 years. | [106,107,108,109] |
| NCT03929601 | Suspended | February 2020 | Ongoing | Interventional | Result not published. | |
| NCT00645840 | Completed | March 2008 | September 2009 | Interventional | Anakinra-treated patients had similar glycated hemoglobin and MMTT responses but lower insulin requirements 1 and 4 months after diagnosis compared with controls and lower insulin-dose-adjusted glycated hemoglobin 1 month after diagnosis. | [116,117] |
| NCT00730392 | Completed | October 2002 | January 2008 | Interventional | Treatment of pediatric patients newly diagnosed with type 1 diabetes with etanercept resulted in lower glycated hemoglobin and increased endogenous insulin production, suggesting the preservation of beta-cell function. | [123] |
| NCT02293837 | Completed | March 2015 | August 2020 | Interventional | Tocilizumab reduced T cell IL-6R signaling but did not modulate CD4+ T cell phenotypes or slow the loss of residual β cell function in newly diagnosed individuals with type 1 diabetes. | [127] |
| NCT02117765 | Unknown | March 2015 | June 2017 | Interventional | Ustekinumab was deemed safe to progress to efficacy studies at doses used to treat psoriasis in adults with T1D. A 90 mg maintenance dosing schedule reduced proinsulin-specific IFN-γ and IL-17A-producing T cells. Further studies are warranted to determine whether Ustekinumab can prevent C-peptide AUC decline and induce a clinical response. | [132] |
| NCT00445913 | Completed | March 2007 | February 2016 | Interventional | Treatment with autologous dendritic cells in a native state or directed ex vivo toward a tolerogenic immunosuppressive state is safe and well-tolerated. Dendritic cells upregulated the frequency of a potentially beneficial B220+ CD11c2 B-cell population, at least in type 1 diabetes autoimmunity. | [153] |
| NCT02354911 | Unknown | October 2015 | January 2019 | Interventional | Result not published | |
| NCT01947569 | Unknown | October 2013 | November 2013 | Interventional | Result not published | |
| NCT04590872 | Recruiting | April 2022 | Ongoing | Interventional | Result not published |
8. Future Views and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patterson, C.; Guariguata, L.; Dahlquist, G.; Soltész, G.; Ogle, G.; Silink, M. Diabetes in the young—A global view and worldwide estimates of numbers of children with type 1 diabetes. Diabetes Res. Clin. Pract. 2014, 103, 161–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, C.C.; Karuranga, S.; Salpea, P.; Saeedi, P.; Dahlquist, G.; Soltesz, G.; Ogle, G.D. Worldwide estimates of incidence, prevalence and mortality of type 1 diabetes in children and adolescents: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilonen, J.; Lempainen, J.; Veijola, R. The heterogeneous pathogenesis of type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 635–650. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Palombi, M.; Fierabracci, A. The putative role of the C1858T polymorphism of protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun. Rev. 2013, 12, 717–725. [Google Scholar] [CrossRef]
- Robertson, C.C.; Rich, S.S. Genetics of type 1 diabetes. Curr. Opin. Genet. Dev. 2018, 50, 7–16. [Google Scholar] [CrossRef]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Münz, C.; Steinman, R.M.; Fujii, S. Dendritic cell maturation by innate lymphocytes: Coordinated stimulation of innate and adaptive immunity. J. Exp. Med. 2005, 202, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Halim, T.Y.F.; Hwang, Y.Y.; Scanlon, S.T.; Zaghouani, H.; Garbi, N.; Fallon, P.G.; McKenzie, A.N.J. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 2016, 17, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Wege, A.K.; Ritter, U. Characteristics of ‘Tip-DCs and MDSCs’ and Their Potential Role in Leishmaniasis. Front. Microbiol. 2012, 3, 74. [Google Scholar] [CrossRef] [Green Version]
- Heath, W.R.; Carbone, F.R. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 2009, 10, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.K.; Narni-Mancinelli, E.; Senechal, B.; Trouillet, C.; Saederup, N.; Leemput, J.; Bigot, K.; Campisi, L.; Abitbol, M.; et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J. Exp. Med. 2009, 206, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Waskow, C.; Liu, K.; Darrasse-Jèze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef]
- Hettinger, J.; Richards, D.M.; Hansson, J.; Barra, M.M.; Joschko, A.-C.; Krijgsveld, J.; Feuerer, M. Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 2013, 14, 821–830. [Google Scholar] [CrossRef]
- Meredith, M.M.; Liu, K.; Darrasse-Jeze, G.; Kamphorst, A.O.; Schreiber, H.A.; Guermonprez, P.; Idoyaga, J.; Cheong, C.; Yao, K.H.; Niec, R.E.; et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 2012, 209, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Cisse, B.; Caton, M.L.; Lehner, M.; Maeda, T.; Scheu, S.; Locksley, R.; Holmberg, D.; Zweier, C.; den Hollander, N.S.; Kant, S.G.; et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 2008, 135, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Schlitzer, A.; McGovern, N.; Teo, P.; Zelante, T.; Atarashi, K.; Low, D.; Ho, A.W.S.; See, P.; Shin, A.; Wasan, P.S.; et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 2013, 38, 970–983. [Google Scholar] [CrossRef] [Green Version]
- Sichien, D.; Scott, C.L.; Martens, L.; Vanderkerken, M.; Van Gassen, S.; Plantinga, M.; Joeris, T.; De Prijck, S.; Vanhoutte, L.; Vanheerswynghels, M.; et al. IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 2016, 45, 626–640. [Google Scholar] [CrossRef] [Green Version]
- Besin, G.; Gaudreau, S.; Dumont-Blanchette, E.; Ménard, M.; Guindi, C.; Dupuis, G.; Amrani, A. IFN regulatory factors 4 and 8 expression in the NOD mouse. Clin. Dev. Immunol. 2011, 2011, 374859. [Google Scholar] [CrossRef] [PubMed]
- Perié, L.; Naik, S.H. Toward defining a ‘lineage’—The case for dendritic cells. Semin. Cell Dev. Biol. 2015, 41, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Proietto, A.I.; Wilson, N.S.; Dakic, A.; Schnorrer, P.; Fuchsberger, M.; Lahoud, M.H.; O’Keeffe, M.; Shao, Q.X.; Chen, W.F.; et al. Cutting edge: Generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 2005, 174, 6592–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkling, M.E.; Cytlak, U.; Lau, C.M.; Lewis, K.L.; Resteu, A.; Khodadadi-Jamayran, A.; Siebel, C.W.; Salmon, H.; Merad, M.; Tsirigos, A.; et al. Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell Rep. 2018, 23, 3658–3672.e6. [Google Scholar] [CrossRef]
- Nobs, S.P.; Schneider, C.; Dietrich, M.G.; Brocker, T.; Rolink, A.; Hirsch, E.; Kopf, M. PI3-Kinase-γ Has a Distinct and Essential Role in Lung-Specific Dendritic Cell Development. Immunity 2015, 43, 674–689. [Google Scholar] [CrossRef] [Green Version]
- McKenna, H.J.; Stocking, K.L.; Miller, R.E.; Brasel, K.; De Smedt, T.; Maraskovsky, E.; Maliszewski, C.R.; Lynch, D.H.; Smith, J.; Pulendran, B.; et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95, 3489–3497. [Google Scholar] [CrossRef]
- Whartenby, K.A.; Calabresi, P.A.; McCadden, E.; Nguyen, B.; Kardian, D.; Wang, T.; Mosse, C.; Pardoll, D.M.; Small, D. Inhibition of FLT3 signaling targets DCs to ameliorate autoimmune disease. Proc. Natl. Acad. Sci. USA 2005, 102, 16741–16746. [Google Scholar] [CrossRef] [Green Version]
- Edelson, B.T.; Bradstreet, T.R.; Kc, W.; Hildner, K.; Herzog, J.W.; Sim, J.; Russell, J.H.; Murphy, T.L.; Unanue, E.R.; Murphy, K.M. Batf3-Dependent CD11b low/− Peripheral Dendritic Cells Are GM-CSF-Independent and Are Not Required for Th Cell Priming after Subcutaneous Immunization. PLoS ONE 2011, 6, e25660. [Google Scholar] [CrossRef]
- Bogunovic, M.; Ginhoux, F.; Helft, J.; Shang, L.; Hashimoto, D.; Greter, M.; Liu, K.; Jakubzick, C.; Ingersoll, M.A.; Leboeuf, M.; et al. Origin of the lamina propria dendritic cell network. Immunity 2009, 31, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Esashi, E.; Wang, Y.H.; Perng, O.; Qin, X.F.; Liu, Y.J.; Watowich, S.S. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 2008, 28, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onai, N.; Obata-Onai, A.; Schmid, M.A.; Ohteki, T.; Jarrossay, D.; Manz, M.G. Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat. Immunol. 2007, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)fining the dendritic cell lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, P.A. Dendritic cell subsets in type 1 diabetes: Friend or foe? Front. Immunol. 2013, 4, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sichien, D.; Lambrecht, B.N.; Guilliams, M.; Scott, C.L. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef]
- Anderson, D.A.; Dutertre, C.-A.; Ginhoux, F.; Murphy, K.M. Genetic models of human and mouse dendritic cell development and function. Nat. Rev. Immunol. 2021, 21, 101–115. [Google Scholar] [CrossRef]
- Moberg, C.L. An appreciation of Ralph Marvin Steinman (1943–2011). J. Exp. Med. 2011, 208, 2337–2342. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Ko, H.-J.; Kweon, M.-N. Mucosal dendritic cells shape mucosal immunity. Exp. Mol. Med. 2014, 46, e84. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.; Martin, S.; Garg, A.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comabella, M.; Montalban, X.; Münz, C.; Lünemann, J.D. Targeting dendritic cells to treat multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watarai, H.; Hinohara, A.; Nagafune, J.; Nakayama, T.; Taniguchi, M.; Yamaguchi, Y. Plasma membrane-focused proteomics: Dramatic changes in surface expression during the maturation of human dendritic cells. Proteomics 2005, 5, 4001–4011. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.-H.; Shin, H.W.; Lee, J.M.; Lee, H.-W.; Kim, E.-C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediat. Inflamm. 2015, 2015, 794143. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.M.; Chen, J.; Yauch, L.E.; Levitz, S.M. Opsonic requirements for dendritic cell-mediated responses to Cryptococcus neoformans. Infect. Immun. 2005, 73, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcγ receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef]
- Hammer, G.E.; Ma, A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu. Rev. Immunol. 2013, 31, 743–791. [Google Scholar] [CrossRef] [Green Version]
- Audiger, C.; Rahman, M.J.; Yun, T.J.; Tarbell, K.V.; Lesage, S. The Importance of Dendritic Cells in Maintaining Immune Tolerance. J. Immunol. 2017, 198, 2223–2231. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Steimle, A.; Frick, J.-S. Molecular Mechanisms of Induction of Tolerant and Tolerogenic Intestinal Dendritic Cells in Mice. J. Immunol. Res. 2016, 2016, 1958650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, O.; DeKruyff, R.H.; Umetsu, D.T. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2001, 2, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.P.; Platt, N.; Wykes, M.; Major, J.R.; Powell, T.J.; Jenkins, C.D.; MacPherson, G.G. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 2000, 191, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Verginis, P.; Li, H.S.; Carayanniotis, G. Tolerogenic semimature dendritic cells suppress experimental autoimmune thyroiditis by activation of thyroglobulin-specific CD4+CD25+ T cells. J. Immunol. 2005, 174, 7433–7439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frick, J.S.; Zahir, N.; Müller, M.; Kahl, F.; Bechtold, O.; Lutz, M.B.; Kirschning, C.J.; Reimann, J.; Jilge, B.; Bohn, E.; et al. Colitogenic and non-colitogenic commensal bacteria differentially trigger DC maturation and Th cell polarization: An important role for IL-6. Eur. J. Immunol. 2006, 36, 1537–1547. [Google Scholar] [CrossRef]
- Jaen, O.; Rullé, S.; Bessis, N.; Zago, A.; Boissier, M.-C.; Falgarone, G. Dendritic cells modulated by innate immunity improve collagen-induced arthritis and induce regulatory T cells in vivo. Immunology 2009, 126, 35–44. [Google Scholar] [CrossRef]
- Yang, X.J.; Meng, S.; Zhu, C.F.; Jiang, H.; Wu, W.X. Semi-mature MyD88-silenced bone marrow dendritic cells prolong the allograft survival in a rat model of intestinal transplantation. Chin. Med. J. 2011, 124, 268–272. [Google Scholar]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [Green Version]
- Sia, J.K.; Bizzell, E.; Madan-Lala, R.; Rengarajan, J. Engaging the CD40-CD40L pathway augments T-helper cell responses and improves control of Mycobacterium tuberculosis infection. PLoS Pathog. 2017, 13, e1006530. [Google Scholar] [CrossRef]
- Curato, C.; Bernshtein, B.; Zupancič, E.; Dufner, A.; Jaitin, D.; Giladi, A.; David, E.; Chappell-Maor, L.; Leshkowitz, D.; Knobeloch, K.-P.; et al. DC Respond to Cognate T Cell Interaction in the Antigen-Challenged Lymph Node. Front. Immunol. 2019, 10, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, C.; Cui, Z.; Sun, X.; Lei, L.; Feng, X.; Sun, C.; Gu, J.; Han, W. Mannan Derivatives Instruct Dendritic Cells to Induce Th1/Th2 Cells Polarization via Differential Mitogen-Activated Protein Kinase Activation. Scand. J. Immunol. 2016, 83, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kim, T.H. Fundamental role of dendritic cells in inducing Th2 responses. Korean J. Intern. Med. 2018, 33, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agalioti, T.; Villablanca, E.J.; Huber, S.; Gagliani, N. TH17 cell plasticity: The role of dendritic cells and molecular mechanisms. J. Autoimmun. 2018, 87, 50–60. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef]
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Reviews. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Hanley, T.M.; Blay Puryear, W.; Gummuluru, S.; Viglianti, G.A. PPARgamma and LXR signaling inhibit dendritic cell-mediated HIV-1 capture and trans-infection. PLoS Pathog. 2010, 6, e1000981. [Google Scholar] [CrossRef]
- Kratchmarov, R.; Viragova, S.; Kim, M.J.; Rothman, N.J.; Liu, K.; Reizis, B.; Reiner, S.L. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol. Cell Biol. 2018, 96, 863–871. [Google Scholar] [CrossRef]
- Du, X.; Wen, J.; Wang, Y.; Karmaus, P.W.F.; Khatamian, A.; Tan, H.; Li, Y.; Guy, C.; Nguyen, T.M.; Dhungana, Y.; et al. Hippo/Mst signalling couples metabolic state and immune function of CD8α+ dendritic cells. Nature 2018, 558, 141–145. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, G.; Zeng, H.; Yang, K.; Lamb, R.F.; Chi, H. Tuberous sclerosis 1 (Tsc1)-dependent metabolic checkpoint controls development of dendritic cells. Proc. Natl. Acad. Sci. USA 2013, 110, E4894–E4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Mascanfroni, I.D.; Yeste, A.; Vieira, S.M.; Burns, E.J.; Patel, B.; Sloma, I.; Wu, Y.; Mayo, L.; Ben-Hamo, R.; Efroni, S.; et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol. 2013, 14, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Sim, W.J.; Ahl, P.J.; Connolly, J.E. Metabolism Is Central to Tolerogenic Dendritic Cell Function. Mediat. Inflamm. 2016, 2016, 2636701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Everts, B.; Pearce, E.J. Metabolic control of dendritic cell activation and function: Recent advances and clinical implications. Front. Immunol. 2014, 5, 203. [Google Scholar] [CrossRef] [Green Version]
- Guak, H.; Al Habyan, S.; Ma, E.H.; Aldossary, H.; Al-Masri, M.; Won, S.Y.; Ying, T.; Fixman, E.D.; Jones, R.G.; McCaffrey, L.M.; et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat. Commun. 2018, 9, 2463. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, T.P.; Peakman, M.; Roep, B.O. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin. Exp. Immunol. 2007, 148, 1–16. [Google Scholar] [CrossRef]
- Lehuen, A.; Diana, J.; Zaccone, P.; Cooke, A. Immune cell crosstalk in type 1 diabetes. Nat. Rev. Immunol. 2010, 10, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Amrani, A.; Verdaguer, J.; Thiessen, S.; Bou, S.; Santamaria, P. IL-1alpha, IL-1beta, and IFN-gamma mark beta cells for Fas-dependent destruction by diabetogenic CD4+ T lymphocytes. J. Clin. Investig. 2000, 105, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnuson, A.M.; Thurber, G.M.; Kohler, R.H.; Weissleder, R.; Mathis, D.; Benoist, C. Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proc. Natl. Acad. Sci. USA 2015, 112, 1511–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta-Iwamura, C.; Tarbell, K.V. Type 1 diabetes genetic susceptibility and dendritic cell function: Potential targets for treatment. J. Leukoc. Biol. 2016, 100, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creusot, R.J.; Postigo-Fernandez, J.; Teteloshvili, N. Altered Function of Antigen-Presenting Cells in Type 1 Diabetes: A Challenge for Antigen-Specific Immunotherapy? Diabetes 2018, 67, 1481–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldison, J.; Wong, F.S. Immune and Pancreatic b Cell Interactions in Type 1 Diabetes. Trends Endocrinol. Metab. 2016, 27, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Unanue, E.R.; Ferris, S.T.; Carrero, J.A. The role of islet antigen presenting cells and the presentation of insulin in the initiation of autoimmune diabetes in the NOD mouse. Immunol. Rev. 2016, 272, 183–201. [Google Scholar] [CrossRef]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef]
- Green, E.A.; Eynon, E.E.; Flavell, R.A. Local Expression of TNFalpha in Neonatal NOD Mice Promotes Diabetes by Enhancing Presentation of Islet Antigens. Immunity 1998, 9, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Karlstad, M.D.; Collier, J.J. Transcription of the gene encoding TNF-α is increased by IL-1β in rat and human islets and β-cell lines. Mol. Immunol. 2014, 62, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Zóka, A.; Műzes, G.; Somogyi, A.; Varga, T.; Szémán, B.; Al-Aissa, Z.; Hadarits, O.; Firneisz, G. Altered Immune Regulation in Type 1 Diabetes. Clin. Dev. Immunol. 2013, 2013, 254874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klementowicz, J.E.; Mahne, A.E.; Spence, A.; Nguyen, V.; Satpathy, A.T.; Murphy, K.M.; Tang, Q. Cutting Edge: Origins, Recruitment, and Regulation of CD11c+ Cells in Inflamed Islets of Autoimmune Diabetes Mice. J. Immunol. 2017, 199, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandor, A.M.; Jacobelli, J.; Friedman, R.S. Immune cell trafficking to the islets during type 1 diabetes. Clin. Exp. Immunol. 2019, 198, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheat, W.; Kupfer, R.; Gutches, D.G.; Rayat, G.R.; Beilke, J.; Scheinman, R.I.; Wegmann, D.R. Increased NF-kappa B activity in B cells and bone marrow-derived dendritic cells from NOD mice. Eur. J. Immunol. 2004, 34, 1395–1404. [Google Scholar] [CrossRef]
- Sen, P.; Bhattacharyya, S.; Wallet, M.; Wong, C.P.; Poligone, B.; Sen, M.; Baldwin, A.S., Jr.; Tisch, R. NF-kappa B hyperactivation has differential effects on the APC function of nonobese diabetic mouse macrophages. J. Immunol. 2003, 170, 1770–1780. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, T.; Bunk, M.; Drexhage, H.A.; Leenen, P.J. Bone marrow precursors of nonobese diabetic mice develop into defective macrophage-like dendritic cells in vitro. J. Immunol. 2004, 173, 4342–4351. [Google Scholar] [CrossRef] [Green Version]
- Welzen-Coppens, J.M.; van Helden-Meeuwsen, C.G.; Drexhage, H.A.; Versnel, M.A. Abnormalities of dendritic cell precursors in the pancreas of the NOD mouse model of diabetes. Eur. J. Immunol. 2012, 42, 186–194. [Google Scholar] [CrossRef]
- Burns, S.O. Dendritic cell defects in primary immunodeficiency disorders. LymphoSign J. 2015, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Bigley, V.; Haniffa, M.; Hambleton, S. Human dendritic cell deficiency: The missing ID? Nat. Rev. Immunol. 2011, 11, 575–583. [Google Scholar] [CrossRef]
- Casqueiro, J.; Casqueiro, J.; Alves, C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian J. Endocrinol. Metab. 2012, 16 (Suppl. 1), S27–S36. [Google Scholar] [CrossRef]
- Knapp, S. Diabetes and Infection: Is There a Link?—A Mini-Review. Gerontology 2013, 59, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Olmo, M.; Araña, M. New Insights into Immunotherapy Strategies for Treating Autoimmune Diabetes. Int. J. Mol. Sci. 2019, 20, 4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Wallace, P.M.; Johnson, J.; Gibson, M.G.; Greene, J.L.; Ledbetter, J.A.; Singh, C.; Tepper, M.A. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 1992, 257, 792–795. [Google Scholar] [CrossRef]
- Genovese, M.C.; Pacheco-Tena, C.; Covarrubias, A.; Leon, G.; Mysler, E.; Keiserman, M.; Valente, R.M.; Nash, P.; Simon-Campos, J.A.; Box, J.; et al. Longterm Safety and Efficacy of Subcutaneous Abatacept in Patients with Rheumatoid Arthritis: 5-year Results from a Phase IIIb Trial. J. Rheumatol. 2018, 45, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Cagnotto, G.; Willim, M.; Nilsson, J.-Å.; Compagno, M.; Jacobsson, L.T.H.; Saevarsdottir, S.; Turesson, C. Abatacept in rheumatoid arthritis: Survival on drug, clinical outcomes, and their predictors—data from a large national quality register. Arthritis Res. Ther. 2020, 22, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Orban, T.; Beam, C.A.; Xu, P.; Moore, K.; Jiang, Q.; Deng, J.; Muller, S.; Gottlieb, P.; Spain, L.; Peakman, M. Reduction in CD4 central memory T-cell subset in costimulation modulator abatacept-treated patients with recent-onset type 1 diabetes is associated with slower C-peptide decline. Diabetes 2014, 63, 3449–3457. [Google Scholar] [CrossRef] [Green Version]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greenbaum, C.J.; Speake, C.; Long, S.A.; Dufort, M.J. B lymphocyte alterations accompany abatacept resistance in new-onset type 1 diabetes. JCI Insight 2019, 4, e126136. [Google Scholar] [CrossRef] [Green Version]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef]
- Lopes, M.; Kutlu, B.; Miani, M.; Bang-Berthelsen, C.H.; Størling, J.; Pociot, F.; Goodman, N.; Hood, L.; Welsh, N.; Bontempi, G.; et al. Temporal profiling of cytokine-induced genes in pancreatic β-cells by meta-analysis and network inference. Genomics 2014, 103, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.M.; Wang, X.; Chen, Y.-G.; Jia, S.; Kaldunski, M.L.; Greenbaum, C.J.; the Type 1 Diabetes TrialNet Canakinumab Study Group; Mandrup-Poulsen, T.; the AIDA Study Group; Hessner, M.J. Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. Eur. J. Immunol. 2016, 46, 1030–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainscough, J.S.; Frank Gerberick, G.; Zahedi-Nejad, M.; Lopez-Castejon, G.; Brough, D.; Kimber, I.; Dearman, R.J. Dendritic cell IL-1α and IL-1β are polyubiquitinated and degraded by the proteasome. J. Biol. Chem. 2014, 289, 35582–35592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef]
- Cucak, H.; Hansen, G.; Vrang, N.; Skarsfeldt, T.; Steiness, E.; Jelsing, J. The IL-1β Receptor Antagonist SER140 Postpones the Onset of Diabetes in Female Nonobese Diabetic Mice. J. Diabetes Res. 2016, 2016, 7484601. [Google Scholar] [CrossRef] [Green Version]
- Sumpter, K.M.; Adhikari, S.; Grishman, E.K.; White, P.C. Preliminary studies related to anti-interleukin-1β therapy in children with newly diagnosed type 1 diabetes. Pediatric Diabetes 2011, 12, 656–667. [Google Scholar] [CrossRef]
- Dinarello, C.A.; van der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Psarras, A.; Antanaviciute, A.; Alase, A.; Carr, I.; Wittmann, M.; Emery, P.; Tsokos, G.C.; Vital, E.M. TNF-α Regulates Human Plasmacytoid Dendritic Cells by Suppressing IFN-α Production and Enhancing T Cell Activation. J. Immunol. 2021, 206, 785–796. [Google Scholar] [CrossRef]
- Xu, J.; Eastman, A.J.; Flaczyk, A.; Neal, L.M.; Zhao, G.; Carolan, J.; Malachowski, A.N.; Stolberg, V.R.; Yosri, M.; Chensue, S.W.; et al. Disruption of Early Tumor Necrosis Factor Alpha Signaling Prevents Classical Activation of Dendritic Cells in Lung-Associated Lymph Nodes and Development of Protective Immunity against Cryptococcal Infection. mBio 2016, 7, e00510-16. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-H.; Sun, S.-C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Joyce, D.; Albanese, C.; Steer, J.; Fu, M.; Bouzahzah, B.; Pestell, R.G. NF-κB and cell-cycle regulation: The cyclin connection. Cytokine Growth Factor Rev. 2001, 12, 73–90. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gaynor, R.B. IκB kinases: Key regulators of the NF-κB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Mastrandrea, L.; Yu, J.; Behrens, T.; Buchlis, J.; Albini, C.; Fourtner, S.; Quattrin, T. Etanercept Treatment in Children With New-Onset Type 1 Diabetes. Pilot randomized, placebo-controlled, double-blind study. Diabetes Care 2009, 32, 1244–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, N.; Oriss, T.; Paglia, M.; Ray, A.; Ray, P. A critical role for IL-6 secretion by dendritic cells promoting Th2 and limiting Th1 response (95.24). J. Immunol. 2007, 178, S181. [Google Scholar]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundhausen, C.; Roth, A.; Whalen, E.; Chen, J.; Schneider, A.; Long, S.A.; Wei, S.; Rawlings, R.; Kinsman, M.; Evanko, S.P.; et al. Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci. Transl. Med. 2016, 8, 356ra119. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, C.J.; Serti, E.; Lambert, K.; Weiner, L.J.; Kanaparthi, S.; Lord, S.; Gitelman, S.E.; Wilson, D.M.; Gaglia, J.L.; Griffin, K.J.; et al. IL-6 receptor blockade does not slow β cell loss in new-onset type 1 diabetes. JCI Insight 2021, 6, e150074. [Google Scholar] [CrossRef]
- Gaudino, S.J.; Kumar, P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 2019, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Ban, Y.; Wei, F.; Ma, X. Regulation of Interleukin-12 Production in Antigen-Presenting Cells. Adv. Exp. Med. Biol. 2016, 941, 117–138. [Google Scholar] [CrossRef]
- Powell, M.D.; Read, K.A.; Sreekumar, B.K.; Jones, D.M.; Oestreich, K.J. IL-12 signaling drives the differentiation and function of a TH1-derived TFH1-like cell population. Sci. Rep. 2019, 9, 13991. [Google Scholar] [CrossRef]
- Yu, S.; Jia, L.; Zhang, Y.; Zhong, J.; Yang, B.; Wu, C. IL-12 induced the generation of IL-21- and IFN-γ-co-expressing poly-functional CD4+ T cells from human naive CD4+ T cells. Cell Cycle 2015, 14, 3362–3372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwaha, A.K.; Chow, S.; Pesenacker, A.M.; Cook, L.; Sun, A.; Long, S.A.; Yang, J.H.M.; Ward-Hartstonge, K.A.; Williams, E.; Domingo-Vila, C.; et al. A phase 1b open-label dose-finding study of ustekinumab in young adults with type 1 diabetes. Immunother. Adv. 2022, 2, ltab022. [Google Scholar] [CrossRef] [PubMed]
- Phillips, B.; Nylander, K.; Harnaha, J.; Machen, J.; Lakomy, R.; Styche, A.; Gillis, K.; Brown, L.; Lafreniere, D.; Gallo, M.; et al. A Microsphere-Based Vaccine Prevents and Reverses New-Onset Autoimmune Diabetes. Diabetes 2008, 57, 1544–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, G.; Geliebter, A.; Babad, J.; Santamaria, P.; Serreze, D.V.; Freeman, G.J.; Tarbell, K.V.; Sharpe, A.; DiLorenzo, T.P. DEC-205-mediated antigen targeting to steady-state dendritic cells induces deletion of diabetogenic CD8⁺ T cells independently of PD-1 and PD-L1. Int. Immunol. 2013, 25, 651–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.S.; Stewart, J.M.; Marshall, G.P.; Carstens, M.R.; Zhang, Y.; Dolgova, N.V.; Xia, C.; Brusko, T.M.; Wasserfall, C.H.; Clare-Salzler, M.J.; et al. Dual-Sized Microparticle System for Generating Suppressive Dendritic Cells Prevents and Reverses Type 1 Diabetes in the Nonobese Diabetic Mouse Model. ACS Biomater. Sci. Eng. 2019, 5, 2631–2646. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, J.; Wang, J.; Zhang, W.; Xu, B.; Xu, X.; Zong, L. Targeted delivery of antigen to intestinal dendritic cells induces oral tolerance and prevents autoimmune diabetes in NOD mice. Diabetologia 2018, 61, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Funda, D.P.; Palová-Jelínková, L.; Goliáš, J.; Kroulíková, Z.; Fajstová, A.; Hudcovic, T.; Špíšek, R. Optimal Tolerogenic Dendritic Cells in Type 1 Diabetes (T1D) Therapy: What Can We Learn From Non-obese Diabetic (NOD) Mouse Models? Front. Immunol. 2019, 10, 967. [Google Scholar] [CrossRef]
- Schülke, S. Induction of Interleukin-10 Producing Dendritic Cells As a Tool to Suppress Allergen-Specific T Helper 2 Responses. Front. Immunol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Zhong, M.; Zhong, C.; Cui, W.; Wang, G.; Zheng, G.; Li, L.; Zhang, J.; Ren, R.; Gao, H.; Wang, T.; et al. Induction of tolerogenic dendritic cells by activated TGF-β/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer 2019, 19, 439. [Google Scholar] [CrossRef]
- Tahaghoghi-Hajghorbani, S.; Ajami, A.; Ghorbanalipoor, S.; Hosseini-khah, Z.; Taghiloo, S.; Khaje-Enayati, P.; Hosseini, V. Protective effect of TSLP and IL-33 cytokines in ulcerative colitis. Autoimmun. Highlights 2019, 10, 1. [Google Scholar] [CrossRef]
- Gaudreau, S.; Guindi, C.; Ménard, M.; Besin, G.; Dupuis, G.; Amrani, A. Granulocyte-macrophage colony-stimulating factor prevents diabetes development in NOD mice by inducing tolerogenic dendritic cells that sustain the suppressive function of CD4+CD25+ regulatory T cells. J. Immunol. 2007, 179, 3638–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Barriuso, J.; Mansilla, M.J.; Naranjo-Gómez, M.; Sánchez-Pla, A.; Quirant-Sánchez, B.; Teniente-Serra, A.; Ramo-Tello, C.; Martínez-Cáceres, E.M. Comparative transcriptomic profile of tolerogenic dendritic cells differentiated with vitamin D3, dexamethasone and rapamycin. Sci. Rep. 2018, 8, 14985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, N.K.; Fitzgerald, H.K.; Malara, A.; Hambly, R.; Sweeney, C.M.; Kirby, B.; Fletcher, J.M.; Dunne, A. Naturally derived Heme-Oxygenase 1 inducers attenuate inflammatory responses in human dendritic cells and T cells: Relevance for psoriasis treatment. Sci. Rep. 2018, 8, 10287. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, T.; Takaki, S. RGMB enhances the suppressive activity of the monomeric secreted form of CTLA-4. Sci. Rep. 2019, 9, 6984. [Google Scholar] [CrossRef] [Green Version]
- Tezuka, H.; Ohteki, T. Regulation of IgA Production by Intestinal Dendritic Cells and Related Cells. Front. Immunol. 2019, 10, 1891. [Google Scholar] [CrossRef]
- Guindi, C.; Ménard, M.; Cloutier, A.; Gaudreau, S.; Besin, G.; Larivée, P.; McDonald, P.P.; Dupuis, G.; Amrani, A. Differential role of NF-κB, ERK1/2 and AP-1 in modulating the immunoregulatory functions of bone marrow-derived dendritic cells from NOD mice. Cell. Immunol. 2012, 272, 259–268. [Google Scholar] [CrossRef]
- Guindi, C.; Cloutier, A.; Gaudreau, S.; Zerif, E.; McDonald, P.P.; Tatsiy, O.; Asselin, C.; Dupuis, G. Role of the p38 MAPK/C/EBPβ Pathway in the Regulation of Phenotype and IL-10 and IL-12 Production by Tolerogenic Bone Marrow-Derived Dendritic Cells. Cells 2018, 7, 256. [Google Scholar] [CrossRef] [Green Version]
- Gaudreau, S.; Guindi, C.; Ménard, M.; Benabdallah, A.; Dupuis, G.; Amrani, A. GM-CSF induces bone marrow precursors of NOD mice to skew into tolerogenic dendritic cells that protect against diabetes. Cell. Immunol. 2010, 265, 31–36. [Google Scholar] [CrossRef]
- Zerif, E.; Maalem, A.; Gaudreau, S.; Guindi, C.; Ramzan, M.; Véroneau, S.; Gris, D.; Stankova, J.; Rola-Pleszczynski, M.; Mourad, W.; et al. Constitutively active Stat5b signaling confers tolerogenic functions to dendritic cells of NOD mice and halts diabetes progression. J. Autoimmun. 2017, 76, 63–74. [Google Scholar] [CrossRef]
- Zerif, E.; Khan, F.U.; Raki, A.A.; Lullier, V.; Gris, D.; Dupuis, G.; Amrani, A. Elucidating the Role of Ezh2 in Tolerogenic Function of NOD Bone Marrow-Derived Dendritic Cells Expressing Constitutively Active Stat5b. Int. J. Mol. Sci 2020, 21, 6453. [Google Scholar] [CrossRef]
- Strauss, L.; Bergmann, C.; Szczepanski, M.; Gooding, W.; Johnson, J.T.; Whiteside, T.L. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin. Cancer Res. 2007, 13, 4345–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machen, J.; Harnaha, J.; Lakomy, R.; Styche, A.; Trucco, M.; Giannoukakis, N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J. Immunol. 2004, 173, 4331–4341. [Google Scholar] [CrossRef] [PubMed]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhadj Ali, M.; Liu, Y.F.; Arif, S.; Tatovic, D.; Shariff, H.; Gibson, V.B.; Yusuf, N.; Baptista, R.; Eichmann, M.; Petrov, N.; et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci. Transl. Med. 2017, 9, eaaf7779. [Google Scholar] [CrossRef] [Green Version]
- Gibson, V.B.; Nikolic, T.; Pearce, V.Q.; Demengeot, J.; Roep, B.O.; Peakman, M. Proinsulin multi-peptide immunotherapy induces antigen-specific regulatory T cells and limits autoimmunity in a humanized model. Clin. Exp. Immunol. 2015, 182, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, T.; Zwaginga, J.J.; Uitbeijerse, B.S.; Woittiez, N.J.; de Koning, E.J.; Aanstoot, H.J.; Roep, B.O. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for type 1 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 470–472. [Google Scholar] [CrossRef]
- Belz, G.T.; Nutt, S.L. Transcriptional programming of the dendritic cell network. Nat. Rev. Immunol. 2012, 12, 101–113. [Google Scholar] [CrossRef]
- Di Caro, V.; Phillips, B.; Engman, C.; Harnaha, J.; Trucco, M.; Giannoukakis, N. Retinoic acid-producing, ex-vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive B lymphocytes. Clin. Exp. Immunol. 2013, 174, 302–317. [Google Scholar] [CrossRef]
- Phillips, B.E.; Garciafigueroa, Y.; Engman, C.; Trucco, M.; Giannoukakis, N. Tolerogenic Dendritic Cells and T-Regulatory Cells at the Clinical Trials Crossroad for the Treatment of Autoimmune Disease; Emphasis on Type 1 Diabetes Therapy. Front. Immunol. 2019, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Coppieters, K.T.; Harrison, L.C.; von Herrath, M.G. Trials in type 1 diabetes: Antigen-specific therapies. Clin. Immunol. 2013, 149, 345–355. [Google Scholar] [CrossRef]


| DC Subset | DC Type | Human | Mouse | Transcriptional | TLR | Antigen | Major |
|---|---|---|---|---|---|---|---|
| Markers | Markers | Factors | Presentation | Cytokines | |||
| pDC | Lymphoid- | CD123+ | CD11b− | TCF4 | 1, 2, 4 | Poor | Type I IFN |
| resident DC | CD303+ | CD11c+ | IRF8 | 6, 7, 8, 9 | |||
| CD304+ | CD45RA+ | E2.2 | |||||
| ILT3+ | SIGLEC-H+ | ||||||
| ILT7+ | CD8α+ | ||||||
| DR6 | CCR7+ | ||||||
| cDC1 | Lymphoid- | CD141+ | CD11b− | BATF3 | 1, 2, 3, 4 | Cross presentation | L-12p70 |
| resident DC | Clec9a+ | CD11c+ | IRF8 | 6, 8, 9, 10 | on MHC-class I | IFN-λ | |
| CADM1+ | CD103+ | ID2 | |||||
| CXR1+ | CD45RA− | NFIL3 | |||||
| BTLA+ | CD8α+ | ||||||
| CD11b− | CXR1+ | ||||||
| cDC2 | Migratory DC | CD11b+ | CD11b+ | IRF4 | 2, 4, 5 | Presentation on | ? |
| CD11c+ | CD11c+ | PU.1 | 6, 7, 8, 9 | MHC-class II | |||
| CD1c+ | CD45RA− | Notch2 | |||||
| SIRPα+ | SIRPα+ | ||||||
| Clec4a+ | CD4+ | ||||||
| Clec10a+ | CD8α− | ||||||
| CX3CR1+ | CX3CR1+ | ||||||
| Monocyte- | Induced by | CD11c+ | CD11b+ | KLF4 | 1, 2, 3 | Cross presentation | TNF/iNOS |
| derived DC | inflammation | CD1a+ | CD11c+ | IRF8 | 4, 5,7, 8 | ||
| CD1c+ | LY6C+ | PU.1 | |||||
| SIRPα+ | CD8α− | ||||||
| CD206+ | CCR2+ | ||||||
| Langerhans Migratory DC | CD1a+ | CD11b+ | ID2 | 1, 2, 3 | Presentation of | IL-10 | |
| cells | CD207+ | CD45RA− | RUNX3 | 5, 6, 10 | self-antigens for | ||
| CD123+ | CD8α− | β-catenin | tolerance induction | ||||
| TROP2+ | CXCL10+ | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, F.U.; Khongorzul, P.; Raki, A.A.; Rajasekaran, A.; Gris, D.; Amrani, A. Dendritic Cells and Their Immunotherapeutic Potential for Treating Type 1 Diabetes. Int. J. Mol. Sci. 2022, 23, 4885. https://doi.org/10.3390/ijms23094885
Khan FU, Khongorzul P, Raki AA, Rajasekaran A, Gris D, Amrani A. Dendritic Cells and Their Immunotherapeutic Potential for Treating Type 1 Diabetes. International Journal of Molecular Sciences. 2022; 23(9):4885. https://doi.org/10.3390/ijms23094885
Chicago/Turabian StyleKhan, Farhan Ullah, Puregmaa Khongorzul, Ahmed Aziz Raki, Ashwini Rajasekaran, Denis Gris, and Abdelaziz Amrani. 2022. "Dendritic Cells and Their Immunotherapeutic Potential for Treating Type 1 Diabetes" International Journal of Molecular Sciences 23, no. 9: 4885. https://doi.org/10.3390/ijms23094885