
An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor


Abstract
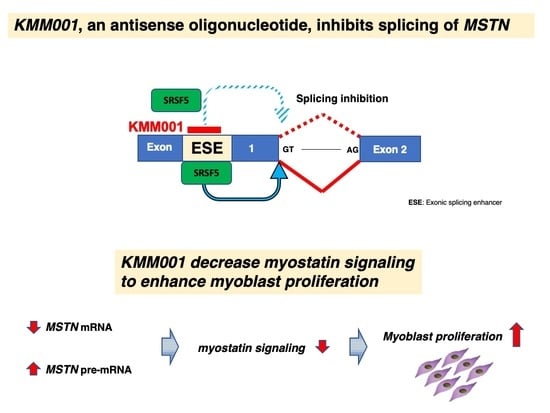
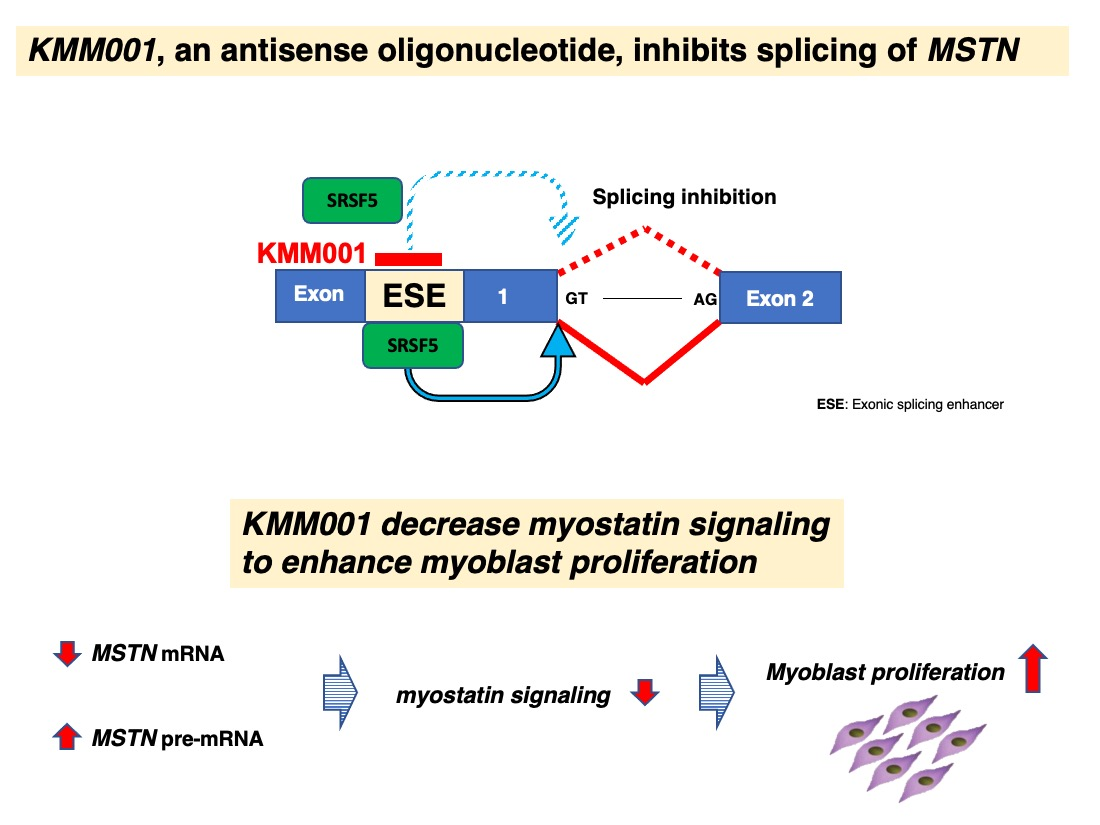
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
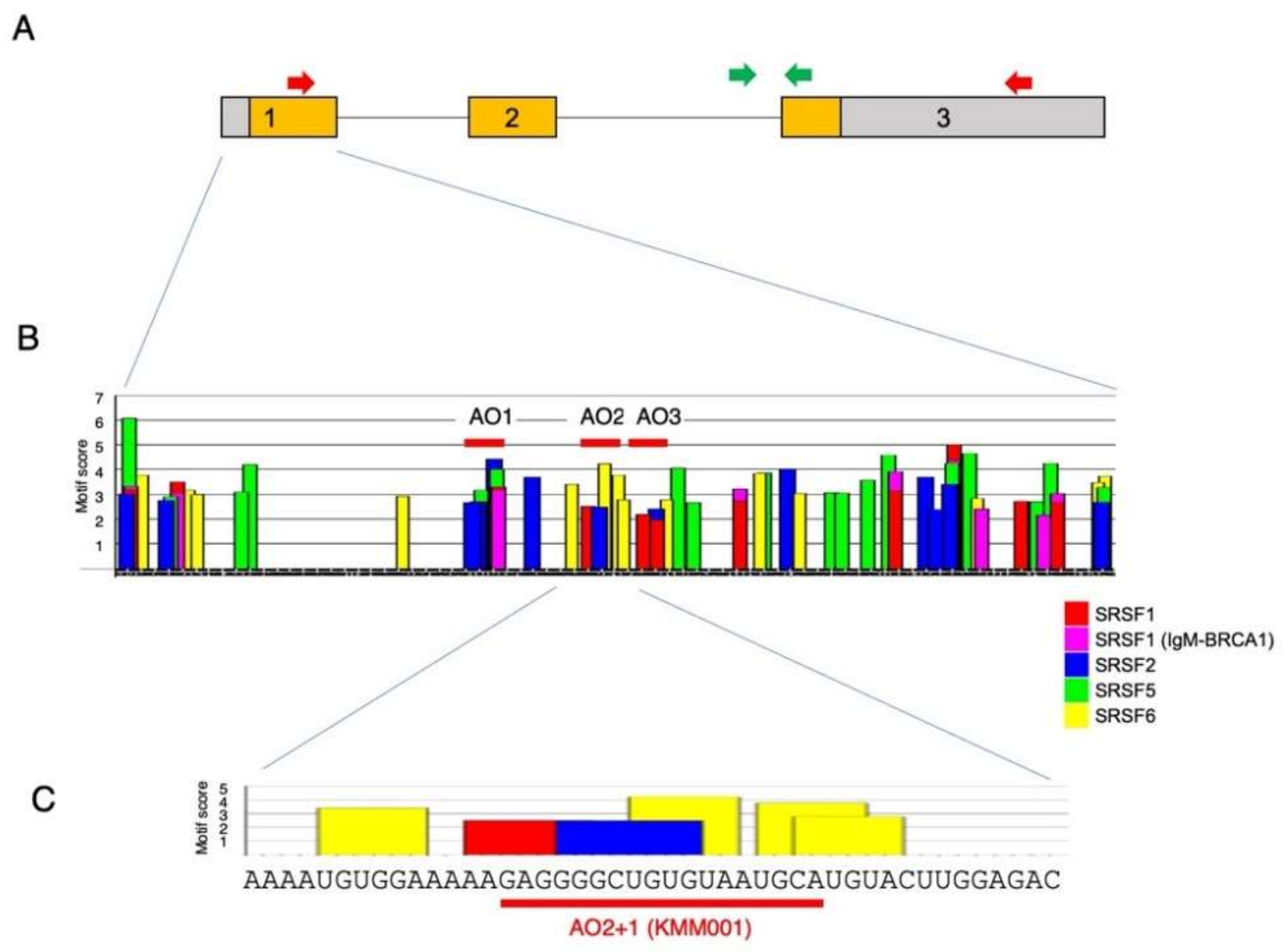
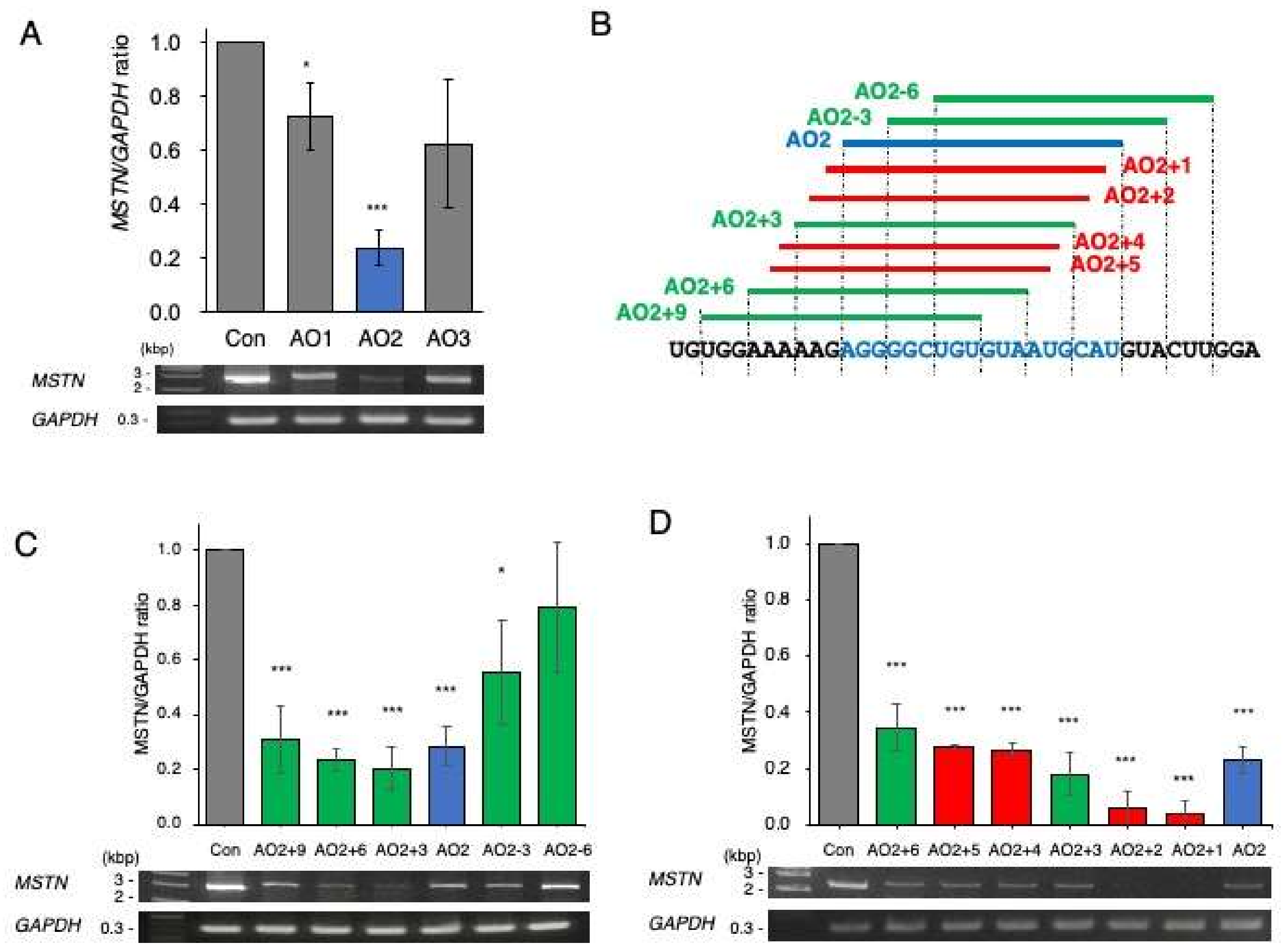
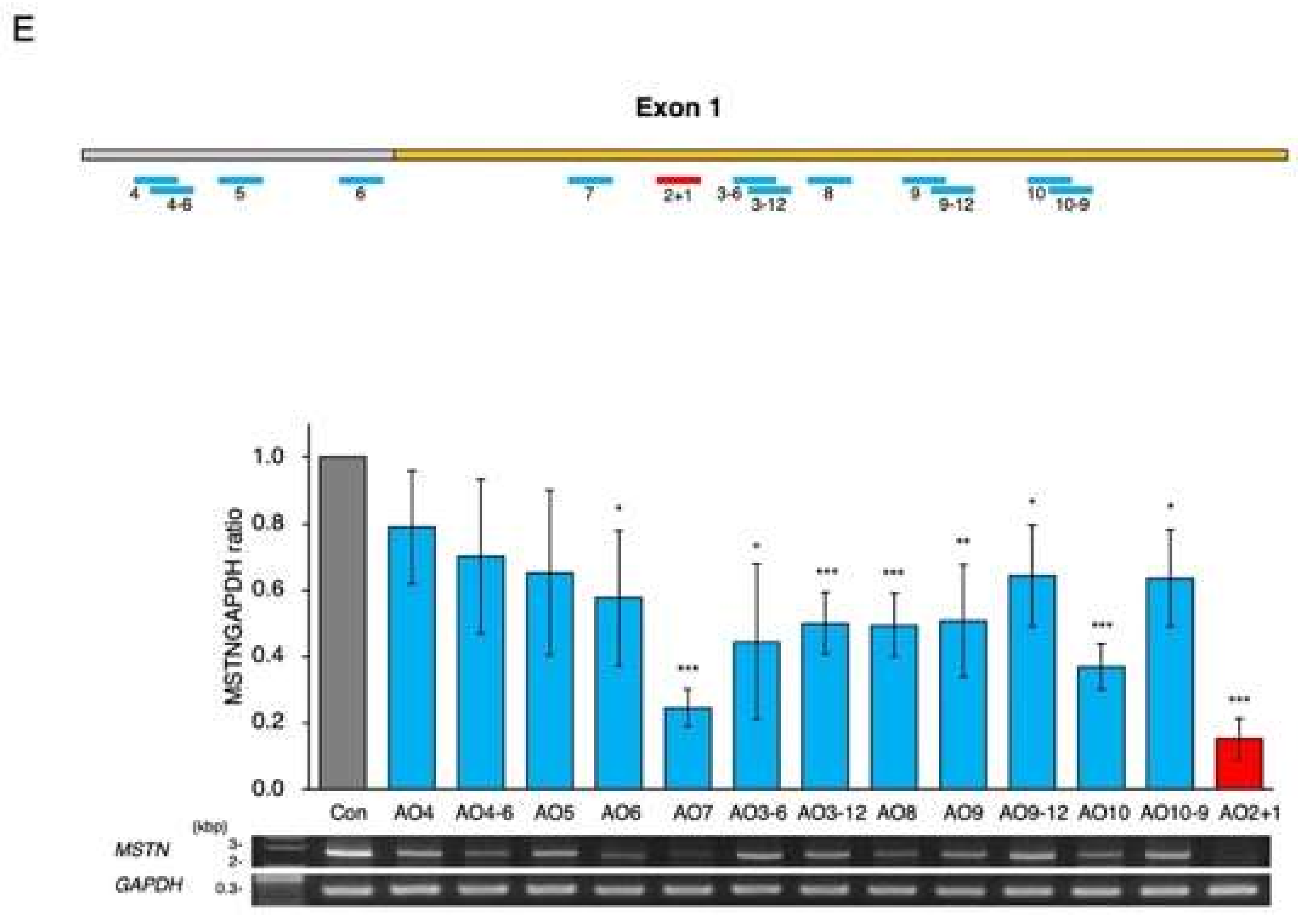
2.1. Identification of a Chimeric ASO That Decreases Mature MSTN mRNA Levels
2.2. Amelioration of AO2+1-Induced Splicing Inhibition by SRSF5
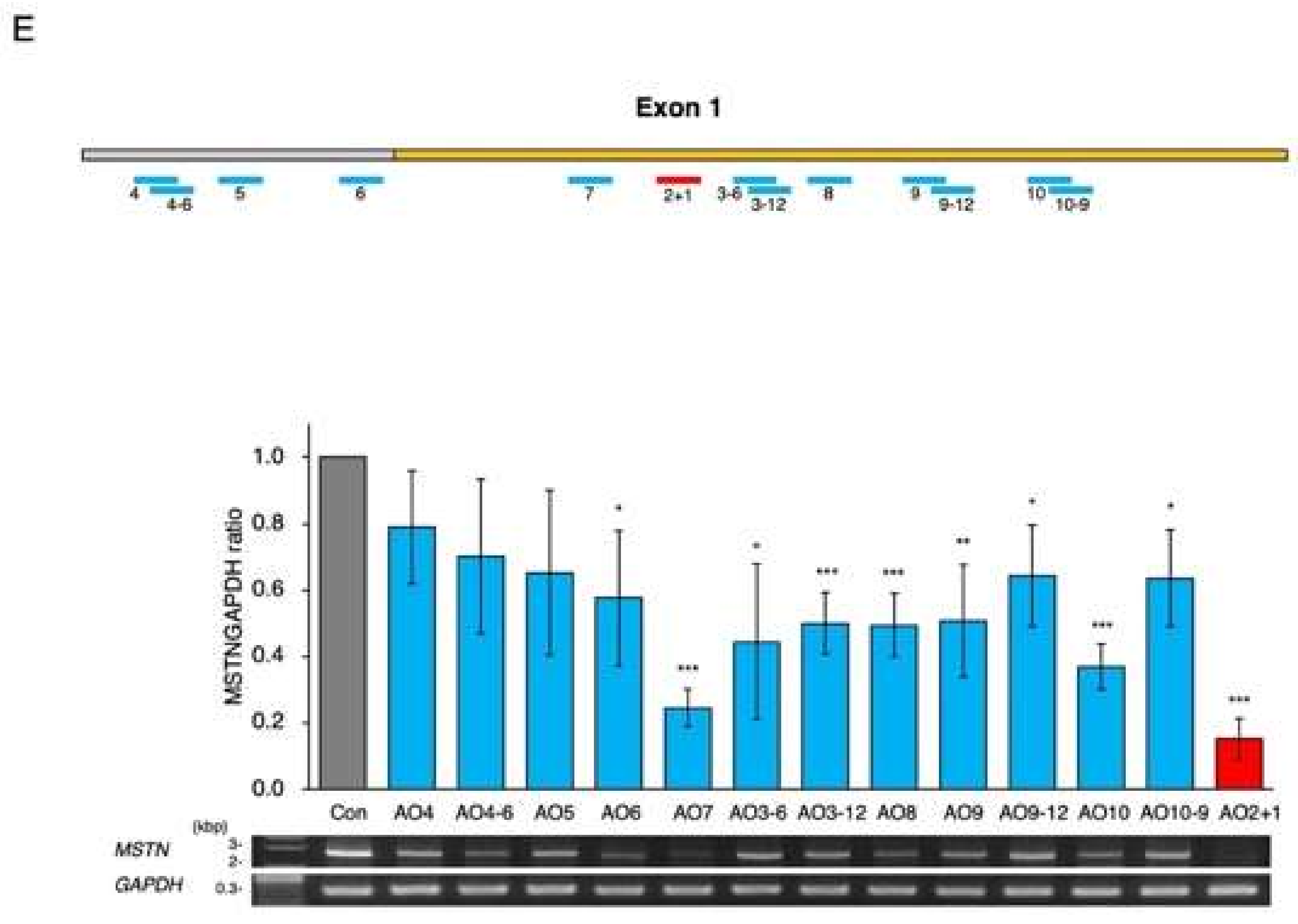
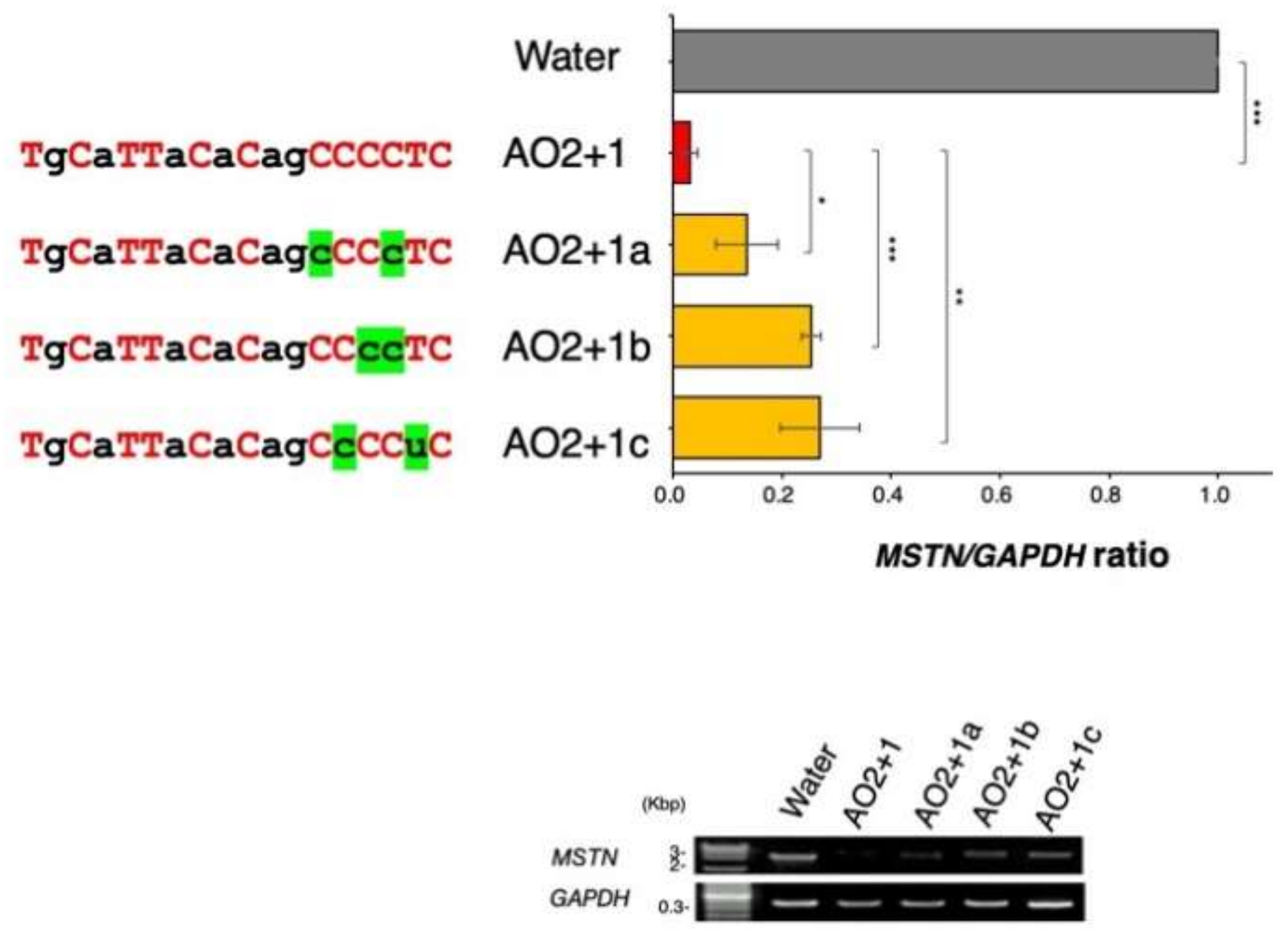
2.3. Determination of the Best Structure of AO2+1
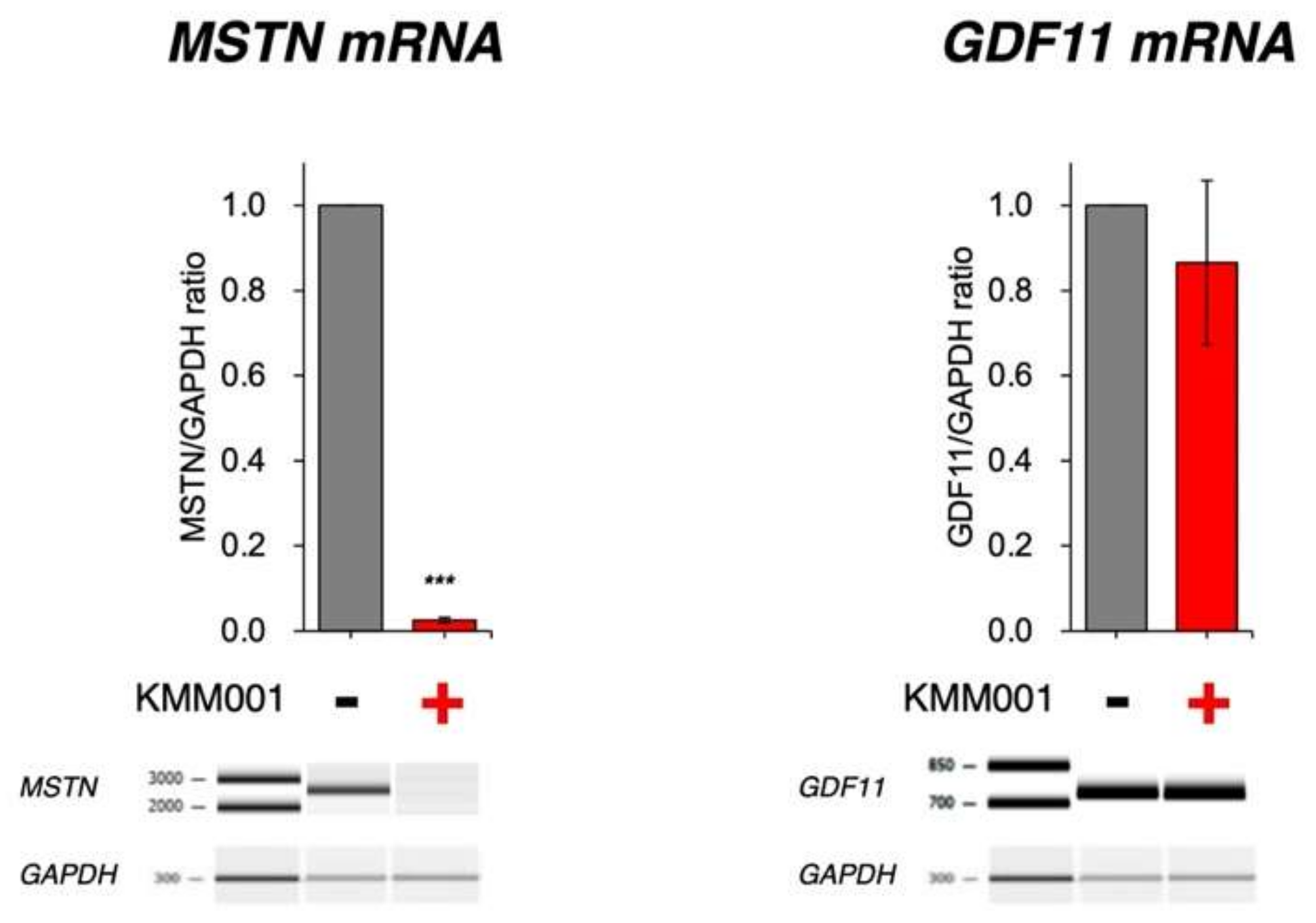
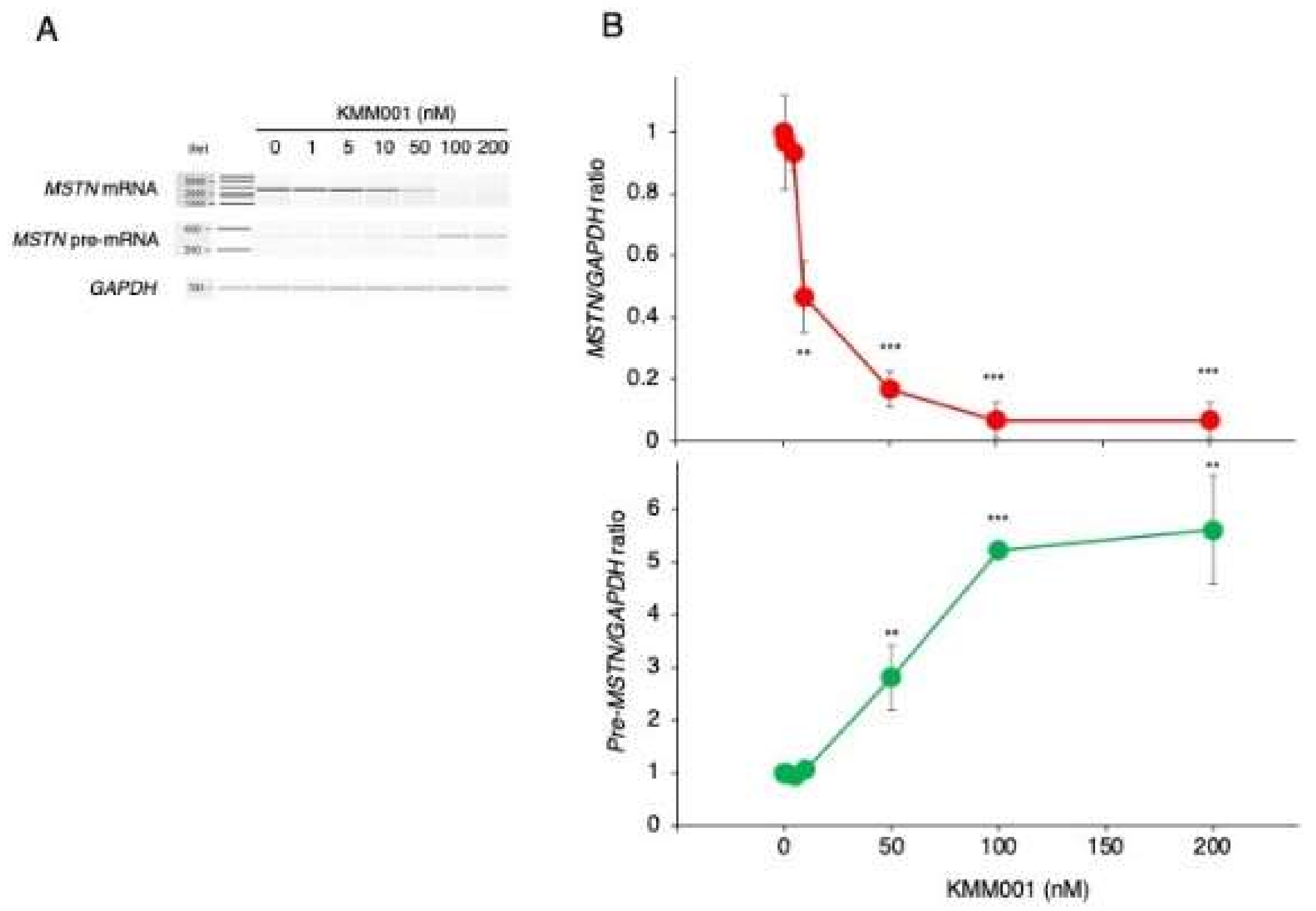
2.4. KMM001 Decreased Mature mRNA Levels in a Dose-Dependent Manner and Reciprocally Increased Pre-mRNA Levels
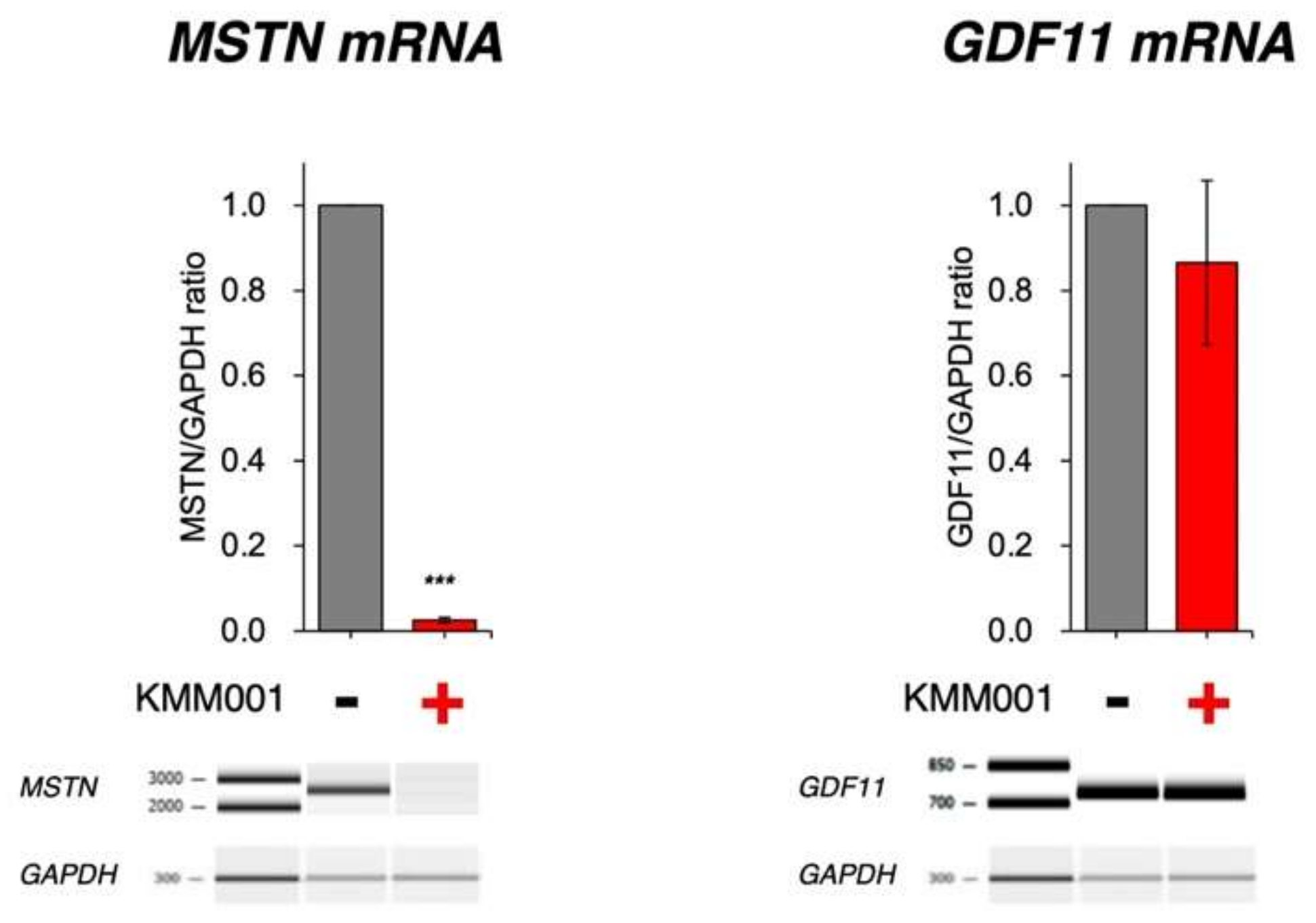
2.5. KMM001 Did Not Decrease GDF11 mRNA Levels
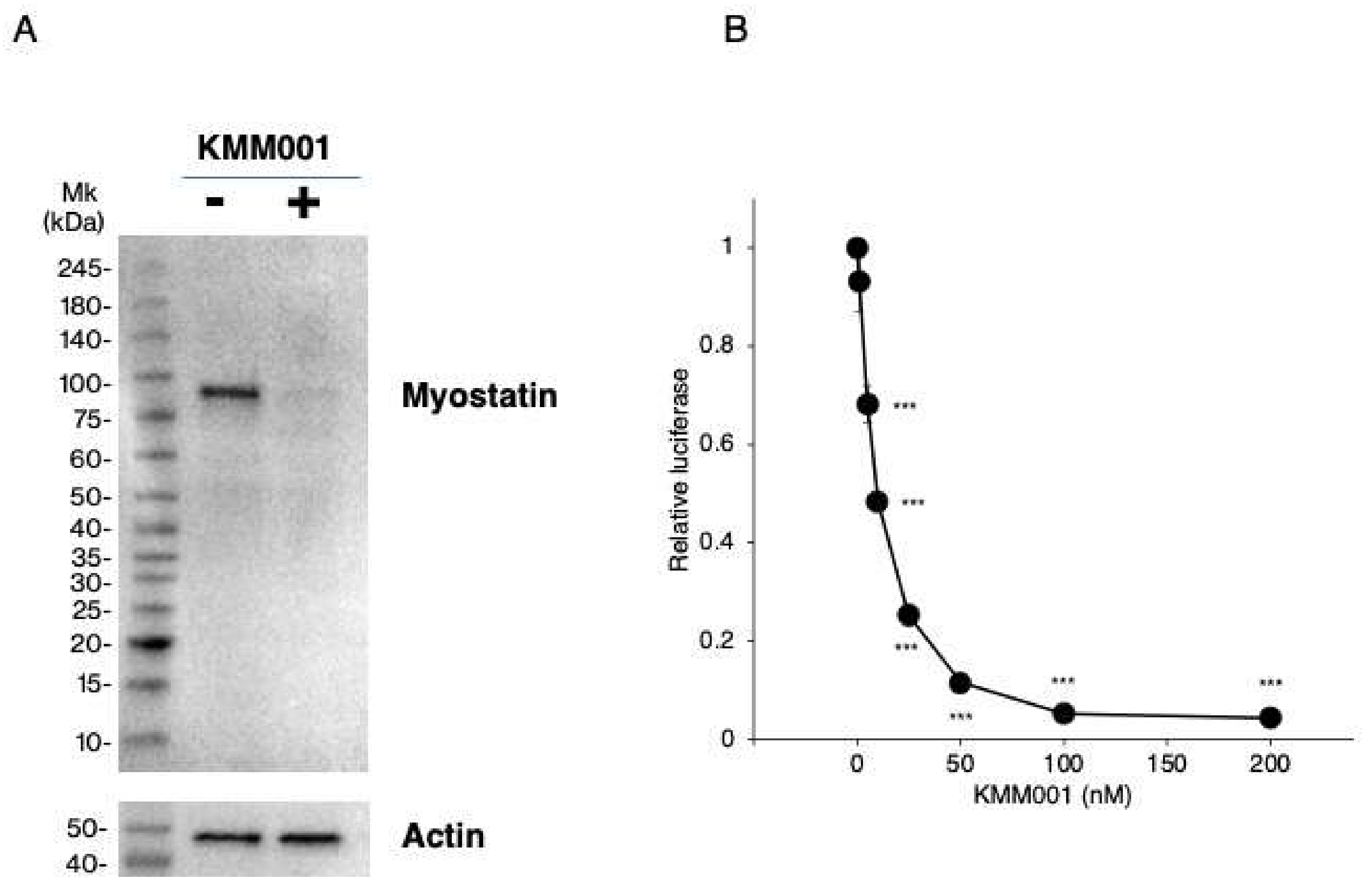
2.6. KMM001 Inhibited Myostatin Protein Expression in CRL-2061 Cells
2.7. KMM001 Inhibited Endogenous Myostatin Signaling in CRL-2061 Cells
2.8. KMM001 Enhanced the Proliferation of Myoblasts
3. Discussion
4. Materials and Methods
4.1. Prediction of Exonic Splicing Enhancer Sequences
4.2. Cells
4.3. Transcript Analysis
4.4. ASOs
4.5. Expression of SRSF5
4.6. Western Blotting
4.7. Myostatin-Signaling Assay with SMAD-Responsive Luciferase
4.8. Cell Proliferation Assays
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Takeshima, Y.; Nishio, H.; Sakamoto, H.; Nakamura, H.; Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin kobe. J. Clin. Investig. 1995, 95, 515–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, M. Duchenne/becker muscular dystrophy: From molecular diagnosis to gene therapy. Brain Dev. 1996, 18, 167–172. [Google Scholar] [CrossRef]
- Pramono, Z.A.; Takeshima, Y.; Alimsardjono, H.; Ishii, A.; Takeda, S.; Matsuo, M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 1996, 226, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Aartsma Rus, A.; Corey, D. The 10th oligonucleotide therapy approved: Golodirsen for duchenne muscular dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from duchenne muscular dystrophy. JMA J. 2021, 4, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.J.A.; Talbot, K.; Bowerman, M. Spinal muscular atrophy: Antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet. 2017, 26, R151–R159. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. Rna mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.-D. Regulation of splicing by sr proteins and sr protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Lee, S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15473835 (accessed on 12 October 2020). [CrossRef]
- Gonzalez-Cadavid, N.F.; Taylor, W.E.; Yarasheski, K.; Sinha-Hikim, I.; Ma, K.; Ezzat, S.; Shen, R.; Lalani, R.; Asa, S.; Mamita, M.; et al. Organization of the human myostatin gene and expression in healthy men and hiv-infected men with muscle wasting. Proc. Natl. Acad. Sci. USA 1998, 95, 14938–14943. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9843994 (accessed on 12 October 2020). [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J. Extracellular regulation of myostatin: A molecular rheostat for muscle mass. Immunol. Endocr. Metab. Agents Med. Chem. 2010, 10, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Langley, B.; Berry, C.; Sharma, M.; Kirk, S.; Bass, J.; Kambadur, R. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J. Biol. Chem. 2000, 275, 40235–40243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9356471 (accessed on 12 October 2020). [CrossRef] [PubMed] [Green Version]
- Grobet, L.; Royo Martin, L.J.; Poncelet, D.; Pirottin, D.; Brouwers, B.; Riquet, J.; Schoeberlein, A.; Dunner, S.; Ménissier, F.; Massabanda, J.; et al. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat. Genet. 1997, 17, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Marcq, F.; Takeda, H.; Pirottin, D.; Tordoir, X.; Bibe, B.; Bouix, J.; Caiment, F.; Elsen, J.M.; Eychenne, F.; et al. A mutation creating a potential illegitimate microrna target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006, 38, 813–818. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16751773 (accessed on 12 October 2020). [CrossRef]
- Mosher, D.; Quignon, P.; Bustamante, C.; Sutter, N.; Mellersh, C.; Parker, H.; Ostrander, E. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007, 3, e79. [Google Scholar] [CrossRef]
- Schuelke, M.; Wagner, K.R.; Stolz, L.E.; Hubner, C.; Riebel, T.; Komen, W.; Braun, T.; Tobin, J.F.; Lee, S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004, 350, 2682–2688. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15215484 (accessed on 12 October 2020). [CrossRef] [Green Version]
- Smith, R.; Lin, B. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support Palliat. Care 2013, 7, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Hoogaars, W.M.H.; Jaspers, R. Past, present, and future perspective of targeting myostatin and related signaling pathways to counteract muscle atrophy. Adv. Exp. Med. Biol. 2018, 1088, 153–206. [Google Scholar] [PubMed]
- Kemaladewi, D.U.; Hoogaars, W.M.; van Heiningen, S.H.; Terlouw, S.; de Gorter, D.J.; den Dunnen, J.T.; van Ommen, G.J.; Aartsma-Rus, A.; Dijke, P.t.; Hoen, P.A.t. Dual exon skipping in myostatin and dystrophin for duchenne muscular dystrophy. BMC Med. Genom. 2011, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilers, W.; Cleasby, M.; Foster, K. Development of antisense-mediated myostatin knockdown for the treatment of insulin resistance. Sci. Rep. 2021, 11, 1604. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.; Debruin, D.; Bagaric, R.; Campelj, D.; Hayes, A. The failed clinical story of myostatin inhibitors against duchenne muscular dystrophy: Exploring the biology behind the battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, Y.; Zhang, H.; Wright, J.; Simmer, J.; Hu, J.C.-C.; Kim, J.-W. Translational attenuation by an intron retention in the 5’ utr of enam causes amelogenesis imperfecta. Biomedicines 2021, 9, 456. [Google Scholar] [CrossRef]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of sma therapy. Hum. Mol. Genet. 2018, 28, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Adachi, H.; Hengesbach, M.; Yu, Y.-T.; Morais, P. From antisense rna to rna modification: Therapeutic potential of rna-based technologies. Biomedicines 2021, 9, 550. [Google Scholar] [CrossRef]
- Lee, T.; Awano, H.; Yagi, M.; Matsumoto, M.; Watanabe, N.; Goda, R.; Koizumi, M.; Takeshima, Y.; Matsuo, M. 2’-o-methyl rna/ethylene-bridged nucleic acid chimera antisense oligonucleotides to induce dystrophin exon 45 skipping. Genes 2017, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Takakusa, H.; Kakuta, M.; Kanda, A.; Takagi, N.; Nagase, H.; Watanabe, N.; Asano, D.; Goda, R.; Masuda, T.; et al. Renadirsen, a novel 2′omerna/ena® chimera antisense oligonucleotide, induces robust exon 45 skipping for dystrophin in vivo. Curr. Issues Mol. Biol. 2021, 43, 90. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.-H.; Lee, K. Muscle hyperplasia in japanese quail by single amino acid deletion in mstn propeptide. Int. J. Mol. Sci. 2020, 21, 1504. [Google Scholar] [CrossRef] [Green Version]
- St Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of duchenne muscular dystrophy and its humanized equivalent, domagrozumab (pf-06252616), increases muscle volume in cynomolgus monkeys. Skelet. Muscle 2017, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Bayarsaikhan, O.; Kawai, N.; Mori, H.; Kinouchi, N.; Nikawa, T.; Tanaka, E. Co-administration of myostatin-targeting sirna and actriib-fc fusion protein increases masseter muscle mass and fiber size. J. Nutr. Sci. Vitaminol. 2017, 63, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, B.; Ward, C. Myostatin/activin receptor ligands in muscle and the development status of attenuating drugs. Endocr. Rev. 2021, 43, 329–365. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, Y.; Takayama, K.; Nishimatsu, S.; Okada, T.; Fujino, M.; Fukai, Y.; Murakami, T.; Hagiwara, H.; Itoh, F.; Tsuchida, K.; et al. The inhibitory core of the myostatin prodomain: Its interaction with both type i and ii membrane receptors, and potential to treat muscle atrophy. PLoS ONE 2015, 10, e0133713. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.; Vissing, J.; Krag, T. Antimyostatin treatment in health and disease: The story of great expectations and limited success. Cells 2021, 10, 533. [Google Scholar] [CrossRef]
- Breitbart, A.; Auger-Messier, M.; Molkentin, J.D.; Heineke, J. Myostatin from the heart: Local and systemic actions in cardiac failure and muscle wasting. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1973–H1982. [Google Scholar] [CrossRef]
- Verzola, D.; Milanesi, S.; Bertolotto, M.; Garibaldi, S.; Villaggio, B.; Brunelli, C.; Balbi, M.; Ameri, P.; Montecucco, F.; Palombo, D.; et al. Myostatin mediates abdominal aortic atherosclerosis progression by inducing vascular smooth muscle cell dysfunction and monocyte recruitment. Sci. Rep. 2017, 7, 46362. [Google Scholar] [CrossRef]
- Coleman, S.K.; Rebalka, I.A.; D’Souza, D.M.; Deodhare, N.; Desjardins, E.M.; Hawke, T.J. Myostatin inhibition therapy for insulin-deficient type 1 diabetes. Sci. Rep. 2016, 6, 32495. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.H.; Bauman, W.A.; Cardozo, C.P. Myostatin inhibits glucose uptake via suppression of insulin-dependent and -independent signaling pathways in myoblasts. Physiol. Rep. 2018, 6, e13837. [Google Scholar] [CrossRef]
- Li, Z.; Kollias, H.; Wagner, K. Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 2008, 283, 19371–19378. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeta, K.; Farea, M.; Nishio, H.; Matsuo, M. An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor. Int. J. Mol. Sci. 2022, 23, 5016. https://doi.org/10.3390/ijms23095016
Maeta K, Farea M, Nishio H, Matsuo M. An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor. International Journal of Molecular Sciences. 2022; 23(9):5016. https://doi.org/10.3390/ijms23095016
Chicago/Turabian StyleMaeta, Kazuhiro, Manal Farea, Hisahide Nishio, and Masafumi Matsuo. 2022. "An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor" International Journal of Molecular Sciences 23, no. 9: 5016. https://doi.org/10.3390/ijms23095016
APA StyleMaeta, K., Farea, M., Nishio, H., & Matsuo, M. (2022). An Antisense Oligonucleotide against a Splicing Enhancer Sequence within Exon 1 of the MSTN Gene Inhibits Pre-mRNA Maturation to Act as a Novel Myostatin Inhibitor. International Journal of Molecular Sciences, 23(9), 5016. https://doi.org/10.3390/ijms23095016