
In Vitro Assays to Identify Metabolism-Disrupting Chemicals with Diabetogenic Activity in a Human Pancreatic β-Cell Model

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
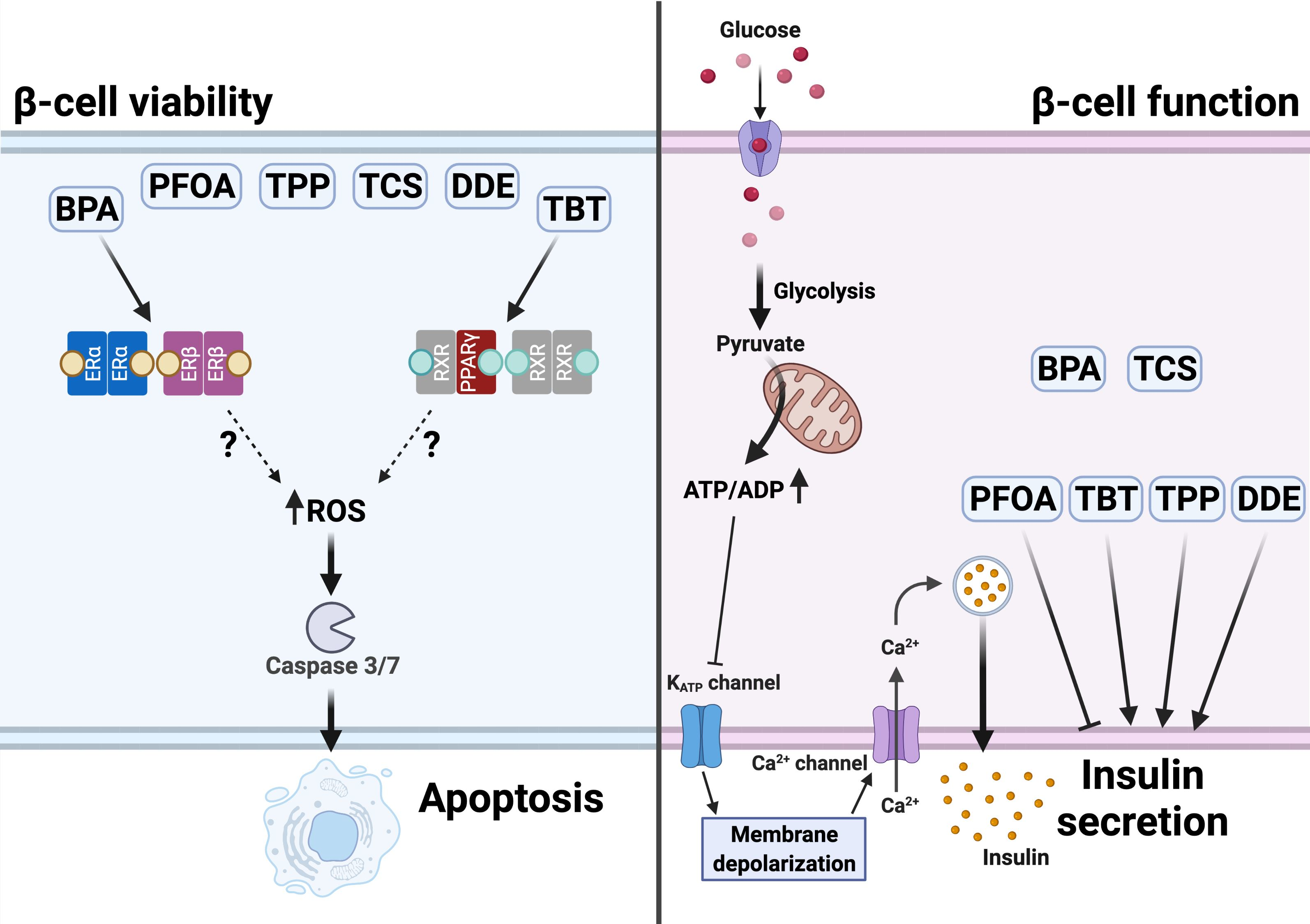
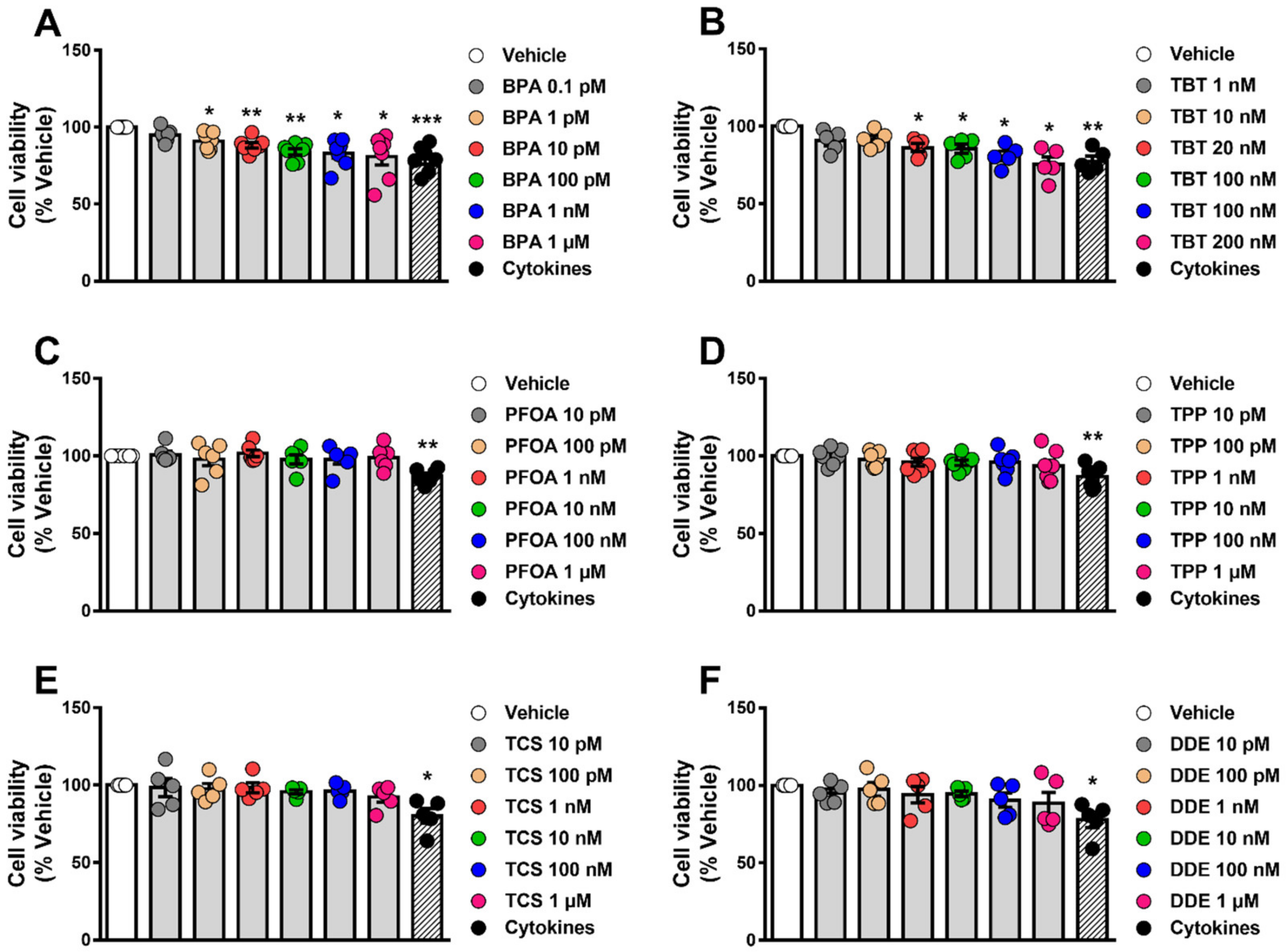
2.1. BPA and TBT Induce β-Cell Death
2.2. Estrogen Receptors Are Involved in BPA-Induced β-Cell Apoptosis
2.3. PPARγ Is Involved in TBT-Induced β-cell Apoptosis
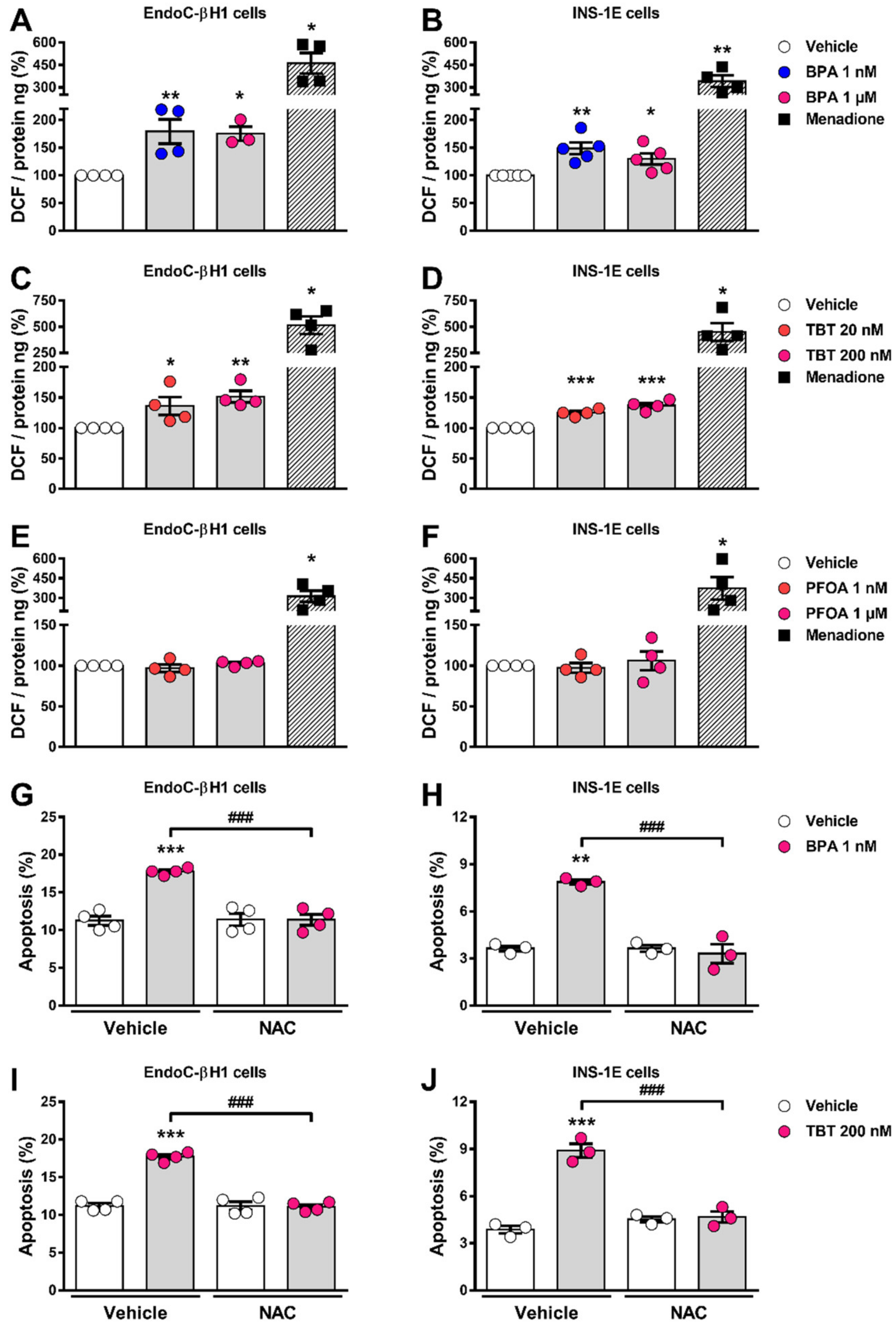
2.4. BPA and TBT, but Not PFOA, Induce ROS Generation
2.5. β-Cell Function Is Disturbed by Different MDCs
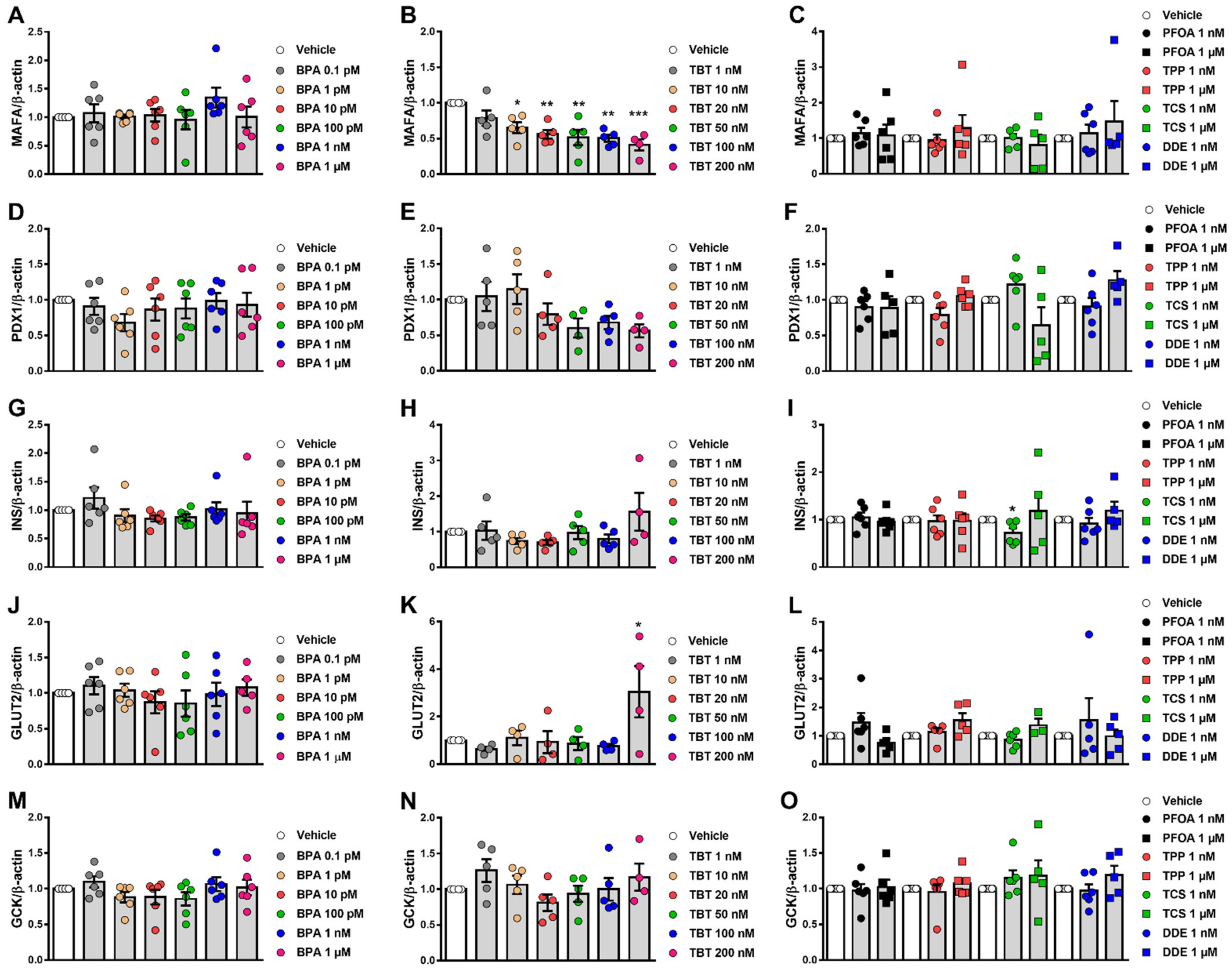
2.6. Effects of MDCs on β-Cell Gene Expression
3. Discussion
3.1. β-Cell Viability Tests
3.2. β-Cell Function Tests
4. Materials and Methods
4.1. Chemicals
4.2. Culture of INS-1E and EndoC-βH1 Cells
4.3. Isolation and Dispersion of Mouse Islets
4.4. Assessment of Cell Viability by MTT Assay
4.5. Assessment of Cell Viability by DNA-Binding Dyes
4.6. Caspase 3/7 Activity Assay
4.7. DCF Assay
4.8. Glucose-Stimulated Insulin Secretion
4.9. RNA Extraction and Real-Time PCR
4.10. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Perez-Serna, A.A.; Babiloni-Chust, I.; Dos Santos, R.S. Type I interferons as key players in pancreatic β-cell dysfunction in type 1 diabetes. Int. Rev. Cell Mol. Biol. 2021, 359, 1–80. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.G. Developmental Exposure to Endocrine Disrupting Chemicals and Type 1 Diabetes Mellitus. Front. Endocrinol. 2018, 9, 513. [Google Scholar] [CrossRef]
- Esser, N.; Utzschneider, K.M.; Kahn, S.E. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia 2020, 63, 2007–2021. [Google Scholar] [CrossRef]
- Corkey, B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes 2012, 61, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 7, 346–353. [Google Scholar] [CrossRef]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [Green Version]
- Mimoto, M.S.; Nadal, A.; Sargis, R.M. Polluted Pathways: Mechanisms of Metabolic Disruption by Endocrine Disrupting Chemicals. Curr. Environ. Health Rep. 2017, 4, 208–222. [Google Scholar] [CrossRef]
- Nadal, A.; Quesada, I.; Tudurí, E.; Nogueiras, R.; Alonso-Magdalena, P. Endocrine-disrupting chemicals and the regulation of energy balance. Nat. Rev. Endocrinol. 2017, 13, 536–546. [Google Scholar] [CrossRef]
- Legler, J.; Zalko, D.; Jourdan, F.; Jacobs, M.; Fromenty, B.; Balaguer, P.; Bourguet, W.; Kos, V.M.; Nadal, A.; Beausoleil, C.; et al. The GOLIATH project: Towards an internationally harmonised approach for testing metabolism disrupting compounds. Int. J. Mol. Sci. 2020, 21, 3480. [Google Scholar] [CrossRef] [PubMed]
- Merglen, A.; Theander, S.; Rubi, B.; Chaffard, G.; Wollheim, C.B.; Maechler, P. Glucose Sensitivity and Metabolism-Secretion Coupling Studied during Two-Year Continuous Culture in INS-1E Insulinoma Cells. Endocrinology 2004, 145, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.E.; Valtat, B.; Bagge, A.; Sharoyko, V.V.; Nicholls, D.G.; Ravassard, P.; Scharfmann, R.; Spégel, P.; Mulder, H. Characterization of Stimulus-Secretion Coupling in the Human Pancreatic EndoC-βH1 Beta Cell Line. PLoS ONE 2015, 10, e0120879. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Chahoud, I.; Heindel, J.J.; Padmanabhan, V.; Paumgartten, F.J.R.; Schoenfelder, G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ. Health Perspect. 2010, 118, 1055–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, K.; Senthilkumar, K.; Giesy, J.P. Occurrence of butyltin compounds in human blood. Environ. Sci. Technol. 1999, 33, 1776–1779. [Google Scholar] [CrossRef]
- Göckener, B.; Weber, T.; Rüdel, H.; Bücking, M.; Kolossa-Gehring, M. Human biomonitoring of per- and polyfluoroalkyl substances in German blood plasma samples from 1982 to 2019. Environ. Int. 2020, 145, 106123. [Google Scholar] [CrossRef]
- Dodson, R.E.; Van Den Eede, N.; Covaci, A.; Perovich, L.J.; Brody, J.G.; Rudel, R.A. Urinary biomonitoring of phosphate flame retardants: Levels in California adults and recommendations for future studies. Environ. Sci. Technol. 2014, 48, 13625–13633. [Google Scholar] [CrossRef] [Green Version]
- Tschersich, C.; Murawski, A.; Schwedler, G.; Rucic, E.; Moos, R.K.; Kasper-Sonnenberg, M.; Koch, H.M.; Brüning, T.; Kolossa-Gehring, M. Bisphenol A and six other environmental phenols in urine of children and adolescents in Germany—Human biomonitoring results of the German Environmental Survey 2014–2017 (GerES V). Sci. Total Environ. 2021, 763, 144615. [Google Scholar] [CrossRef]
- Henríquez-Hernández, L.A.; Ortiz-Andrelluchi, A.; Álvarez-Pérez, J.; Acosta-Dacal, A.; Zumbado, M.; Martínez-González, M.A.; Boada, L.D.; Salas-Salvadó, J.; Luzardo, O.P.; Serra-Majem, L. Human biomonitoring of persistent organic pollutants in elderly people from the Canary Islands (Spain): A temporal trend analysis from the PREDIMED and PREDIMED-Plus cohorts. Sci. Total Environ. 2021, 758, 143637. [Google Scholar] [CrossRef]
- Carchia, E.; Porreca, I.; Almeida, P.J.; D’Angelo, F.; Cuomo, D.; Ceccarelli, M.; De Felice, M.; Mallardo, M.; Ambrosino, C. Evaluation of low doses BPA-induced perturbation of glycemia by toxicogenomics points to a primary role of pancreatic islets and to the mechanism of toxicity. Cell Death Dis. 2015, 6, e1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.F.; Yang, C.Y.; Tsai, J.R.; Wu, C.T.; Liu, S.H.; Lan, K.C. Low-dose tributyltin exposure induces an oxidative stress-triggered JNK-related pancreatic β-cell apoptosis and a reversible hypoinsulinemic hyperglycemia in mice. Sci. Rep. 2018, 8, 5734. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Ropero, A.B.; Carrera, M.P.; Cederroth, C.R.; Baquié, M.; Gauthier, B.R.; Nef, S.; Stefani, E.; Nadal, A. Pancreatic insulin content regulation by the Estrogen receptor ERα. PLoS ONE 2008, 3, e2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroqui, L.; Martinez-Pinna, J.; Castellano-Muñoz, M.; dos Santos, R.S.; Medina-Gali, R.M.; Soriano, S.; Quesada, I.; Gustafsson, J.A.; Encinar, J.A.; Nadal, A. Bisphenol-S and Bisphenol-F alter mouse pancreatic β-cell ion channel expression and activity and insulin release through an estrogen receptor ERβ mediated pathway. Chemosphere 2021, 265, 129051. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, X.; Qiu, L.; Wei, J.; Huang, Q.; Fang, C.; Ye, T.; Kang, M.; Shen, H.; Dong, S. Exposure to bisphenol A induces dysfunction of insulin secretion and apoptosis through the damage of mitochondria in rat insulinoma (INS-1) cells. Cell Death Dis. 2013, 4, e460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hectors, T.L.M.; Vanparys, C.; Pereira-Fernandes, A.; Martens, G.A.; Blust, R. Evaluation of the INS-1 832/13 Cell Line as a Beta-Cell Based Screening System to Assess Pollutant Effects on Beta-Cell Function. PLoS ONE 2013, 8, e60030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaemmaleki, F.; Mohammadi, P.; Baeeri, M.; Navaei-Nigjeh, M.; Abdollahi, M.; Mostafalou, S. Estrogens counteract tributyltin-induced toxicity in the rat islets of Langerhans. Heliyon 2020, 6, e03562. [Google Scholar] [CrossRef]
- Soriano, S.; Alonso-Magdalena, P.; García-Arévalo, M.; Novials, A.; Muhammed, S.J.; Salehi, A.; Gustafsson, J.A.; Quesada, I.; Nadal, A. Rapid insulinotropic action of low doses of Bisphenol-A on mouse and human islets of Langerhans: Role of estrogen receptor β. PLoS ONE 2012, 7, e31109. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Lan, K.C.; Tsai, J.R.; Weng, T.I.; Yang, C.Y.; Liu, S.H. Tributyltin exposure at noncytotoxic doses dysregulates pancreatic β-cell function in vitro and in vivo. Arch. Toxicol. 2017, 91, 3135–3144. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Hoorens, A.; Van De Casteele, M.; Klöppel, G.; Pipeleers, D. Glucose promotes survival of rat pancreatic β cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J. Clin. Investig. 1996, 98, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Babiloni-Chust, I.; dos Santos, R.S.; Medina-Gali, R.M.; Perez-Serna, A.A.; Encinar, J.-A.; Martinez-Pinna, J.; Gustafsson, J.-A.; Marroqui, L.; Nadal, A. G protein-coupled estrogen receptor activation by bisphenol-A disrupts the protection from apoptosis conferred by the estrogen receptors ERα and ERβ in pancreatic beta cells. Environ. Int. 2022, 164, 107250. [Google Scholar] [CrossRef] [PubMed]
- Grün, F.; Blumberg, B. Environmental Obesogens: Organotins and Endocrine Disruption via Nuclear Receptor Signaling. Endocrinology 2006, 147, s50–s55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Maire, A.; Grimaldi, M.; Roecklin, D.; Dagnino, S.; Vivat-Hannah, V.; Balaguer, P.; Bourguet, W. Activation of RXR–PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep. 2009, 10, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA Is a Key Regulator of Glucose-Stimulated Insulin Secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2007, 50, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.-C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2011, 54, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Hang, Y.; Stein, R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol. Metab. 2011, 22, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 371, 606–609. [Google Scholar] [CrossRef]
- Waeber, G.; Thompson, N.; Nicod, P.; Bonny, C. Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol. Endocrinol. 1996, 10, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Watada, H.; Kajimoto, Y.; Umayahara, Y.; Matsuoka, T.; Kaneto, H.; Fujitani, Y.; Kamada, T.; Kawamori, R.; Yamasaki, Y. The Human Glucokinase Gene β-Cell-Type Promoter: An Essential Role of Insulin Promoter Factor 1/PDX-1 in Its Activation in HIT-T15 Cells. Diabetes 1996, 45, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Gannon, M.; Tweedie Ables, E.; Crawford, L.; Lowe, D.; Offield, M.F.; Magnuson, M.A.; Wright, C.V.E. pdx-1 function is specifically required in embryonic β cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev. Biol. 2008, 314, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 Maintains β Cell Identity and Function by Repressing an α Cell Program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Omar-Hmeadi, M.; Idevall-Hagren, O. Insulin granule biogenesis and exocytosis. Cell. Mol. Life Sci. 2021, 78, 1957–1970. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Sternisha, S.M.; Miller, B.G. Molecular and cellular regulation of human glucokinase. Arch. Biochem. Biophys. 2019, 663, 199–213. [Google Scholar] [CrossRef]
- Scharfmann, R.; Staels, W.; Albagli, O. The supply chain of human pancreatic β cell lines. J. Clin. Investig. 2019, 129, 3511–3520. [Google Scholar] [CrossRef] [Green Version]
- Scharfmann, R.; Didiesheim, M.; Richards, P.; Chandra, V.; Oshima, M.; Albagli, O. Mass production of functional human pancreatic β-cells: Why and how? Diabetes Obes. Metab. 2016, 18, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, N.; Márquez, E.J.; Orchard, P.; Narisu, N.; Shamim, M.S.; Thibodeau, A.; Varshney, A.; Kursawe, R.; Erdos, M.R.; Kanke, M.; et al. Multiomic Profiling Identifies cis-Regulatory Networks Underlying Human Pancreatic β Cell Identity and Function. Cell Rep. 2019, 26, 788–801.e6. [Google Scholar] [CrossRef] [Green Version]
- Ryaboshapkina, M.; Saitoski, K.; Hamza, G.M.; Jarnuczak, A.F.; Berthault, C.; Sengupta, K.; Underwood, C.R.; Andersson, S.; Scharfmann, R. Characterization of the secretome, transcriptome and proteome of human β cell line EndoC-βH1. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Gurgul-Convey, E.; Kaminski, M.T.; Lenzen, S. Physiological characterization of the human EndoC-βH1 β-cell line. Biochem. Biophys. Res. Commun. 2015, 464, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hastoy, B.; Godazgar, M.; Clark, A.; Nylander, V.; Spiliotis, I.; van de Bunt, M.; Chibalina, M.V.; Barrett, A.; Burrows, C.; Tarasov, A.I.; et al. Electrophysiological properties of human beta-cell lines EndoC-βH1 and -βH2 conform with human beta-cells. Sci. Rep. 2018, 8, 16994. [Google Scholar] [CrossRef] [PubMed]
- Tsonkova, V.G.; Sand, F.W.; Wolf, X.A.; Grunnet, L.G.; Ringgaard, A.K.; Ingvorsen, C.; Winkel, L.; Kalisz, M.; Dalgaard, K.; Bruun, C.; et al. The EndoC-βH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol. Metab. 2018, 8, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Rutti, S.; Sauter, N.S.; Bouzakri, K.; Prazak, R.; Halban, P.A.; Donath, M.Y. In Vitro Proliferation of Adult Human Beta-Cells. PLoS One 2012, 7, e35801. [Google Scholar] [CrossRef] [PubMed]
- Diedisheim, M.; Oshima, M.; Albagli, O.; Huldt, C.W.; Ahlstedt, I.; Clausen, M.; Menon, S.; Aivazidis, A.; Andreasson, A.C.; Haynes, W.G.; et al. Modeling human pancreatic beta cell dedifferentiation. Mol. Metab. 2018, 10, 74–86. [Google Scholar] [CrossRef]
- Colli, M.L.; Ramos-Rodríguez, M.; Nakayasu, E.S.; Alvelos, M.I.; Lopes, M.; Hill, J.L.E.; Turatsinze, J.V.; Coomans de Brachène, A.; Russell, M.A.; Raurell-Vila, H.; et al. An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat. Commun. 2020, 11, 2584. [Google Scholar] [CrossRef]
- Marroqui, L.; Santin, I.; Dos Santos, R.S.; Marselli, L.; Marchetti, P.; Eizirik, D.L. BACH2, a Candidate Risk Gene for Type 1 Diabetes, Regulates Apoptosis in Pancreatic β-Cells via JNK1 Modulation and Crosstalk with the Candidate Gene PTPN2. Diabetes 2014, 63, 2516–2527. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, R.S.; Marroqui, L.; Grieco, F.A.; Marselli, L.; Suleiman, M.; Henz, S.R.; Marchetti, P.; Wernersson, R.; Eizirik, D.L. Protective role of complement C3 against cytokine-mediated β-cell apoptosis. Endocrinology 2017, 158, 2503–2521. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Sandler, S.; Welsh, N.; Cetkovic-Cvrlje, M.; Nieman, A.; Geller, D.A.; Pipeleers, D.G.; Bendtzen, K.; Hellerström, C. Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J. Clin. Invest. 1994, 93, 1968–1974. [Google Scholar] [CrossRef] [Green Version]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Magueresse-Battistoni, B.; Multigner, L.; Beausoleil, C.; Rousselle, C. Effects of bisphenol A on metabolism and evidences of a mode of action mediated through endocrine disruption. Mol. Cell. Endocrinol. 2018, 475, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Lin, Y.; Li, Y.; Ying, C.; Chen, J.; Song, L.; Zhou, Z.; Lv, Z.; Xia, W.; Chen, X.; et al. Perinatal Exposure to Bisphenol A at Reference Dose Predisposes Offspring to Metabolic Syndrome in Adult Rats on a High-Fat Diet. Endocrinology 2011, 152, 3049–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, A.; Rashid, C.; Xin, F.; Li, C.; Polyak, E.; Duemler, A.; van der Meer, T.; Stefaniak, M.; Wajid, S.; Doliba, N.; et al. Sex- and dose-specific effects of maternal bisphenol A exposure on pancreatic islets of first- and second-generation adult mice offspring. Environ. Health Perspect. 2017, 125, 097022. [Google Scholar] [CrossRef] [PubMed]
- Babiloni-Chust, I.; dos Santos, R.S.; Medina-Gali, R.M.; Perez-Serna, A.A.; Encinar, J.-A.; Martinez-Pinna, J.; Gustafsson, J.-A.; Marroqui, L.; Nadal, A. G protein-coupled estrogen receptor activation by Bisphenol-A disrupts protection from apoptosis conferred by estrogen receptors ERα and ERβ in pancreatic beta cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zuo, Z.; Wu, T.; Lin, M.; Zhang, S.; Yan, F.; Yang, Z.; Wang, Y.; Wang, C. Chronic exposure to tributyltin chloride induces pancreatic islet cell apoptosis and disrupts glucose homeostasis in male mice. Environ. Sci. Technol. 2014, 48, 5179–5186. [Google Scholar] [CrossRef]
- Suh, K.S.; Choi, E.M.; Kim, Y.J.; Hong, S.M.; Park, S.Y.; Rhee, S.Y.; Oh, S.; Kim, S.W.; Pak, Y.K.; Choe, W.; et al. Perfluorooctanoic acid induces oxidative damage and mitochondrial dysfunction in pancreatic β-cells. Mol. Med. Rep. 2017, 15, 3871–3878. [Google Scholar] [CrossRef] [Green Version]
- Ajao, C.; Andersson, M.A.; Teplova, V.V.; Nagy, S.; Gahmberg, C.G.; Andersson, L.C.; Hautaniemi, M.; Kakasi, B.; Roivainen, M.; Salkinoja-Salonen, M. Mitochondrial toxicity of triclosan on mammalian cells. Toxicol. Rep. 2015, 2, 624–637. [Google Scholar] [CrossRef] [Green Version]
- Bowen, C.; Childers, G.; Perry, C.; Martin, N.; McPherson, C.A.; Lauten, T.; Santos, J.; Harry, G.J. Mitochondrial-related effects of pentabromophenol, tetrabromobisphenol A, and triphenyl phosphate on murine BV-2 microglia cells. Chemosphere 2020, 255, 126919. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, J.; Shi, M.; Guo, L.; Liu, L.; Tang, H.; Liu, X. Triphenyl phosphate disturbs the lipidome and induces endoplasmic reticulum stress and apoptosis in JEG-3 cells. Chemosphere 2021, 275, 129978. [Google Scholar] [CrossRef]
- Wang, X.; Li, F.; Liu, J.; Li, Q.; Ji, C.; Wu, H. New insights into the mechanism of hepatocyte apoptosis induced by typical organophosphate ester: An integrated in vitro and in silico approach. Ecotoxicol. Environ. Saf. 2021, 219, 112342. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liang, X.; Hu, Y.; Wang, Y.; Yu, H.; Yang, K. p,p′-DDE induces mitochondria-mediated apoptosis of cultured rat Sertoli cells. Toxicology 2008, 253, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Song, Y.; Wang, Y.; Liang, X.; Hu, Y.; Guan, X.; Cheng, J.; Yang, K. p,p′-DDE Induces Apoptosis of Rat Sertoli Cells via a FasL-Dependent Pathway. J. Biomed. Biotechnol. 2009, 2009, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlíková, N.; Daniel, P.; Šrámek, J.; Jelínek, M.; Šrámková, V.; Němcová, V.; Balušíková, K.; Halada, P.; Kovář, J. Upregulation of vitamin D-binding protein is associated with changes in insulin production in pancreatic beta-cells exposed to p,p′-DDT and p,p′-DDE. Sci. Rep. 2019, 9, 18026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.P.; Cao, L.Y.; Li, C.H.; Guo, L.H.; Colbourne, J.; Ren, X.M. Perfluoroalkyl Substances Stimulate Insulin Secretion by Islet β Cells via G Protein-Coupled Receptor 40. Environ. Sci. Technol. 2020, 54, 3428–3436. [Google Scholar] [CrossRef]
- Zheng, F.; Sheng, N.; Zhang, H.; Yan, S.; Zhang, J.; Wang, J. Perfluorooctanoic acid exposure disturbs glucose metabolism in mouse liver. Toxicol. Appl. Pharmacol. 2017, 335, 41–48. [Google Scholar] [CrossRef]
- Wu, X.; Xie, G.; Xu, X.; Wu, W.; Yang, B. Adverse bioeffect of perfluorooctanoic acid on liver metabolic function in mice. Environ. Sci. Pollut. Res. 2018, 25, 4787–4793. [Google Scholar] [CrossRef]
- Hines, E.P.; White, S.S.; Stanko, J.P.; Gibbs-Flournoy, E.A.; Lau, C.; Fenton, S.E. Phenotypic dichotomy following developmental exposure to perfluorooctanoic acid (PFOA) in female CD-1 mice: Low doses induce elevated serum leptin and insulin, and overweight in mid-life. Mol. Cell. Endocrinol. 2009, 304, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Pinna, J.; Marroqui, L.; Hmadcha, A.; Lopez-Beas, J.; Soriano, S.; Villar-Pazos, S.; Alonso-Magdalena, P.; Dos Santos, R.S.; Quesada, I.; Martin, F.; et al. Oestrogen receptor β mediates the actions of bisphenol-A on ion channel expression in mouse pancreatic beta cells. Diabetologia 2019, 62, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Villar-Pazos, S.; Martinez-Pinna, J.; Castellano-Muñoz, M.; Alonso-Magdalena, P.; Marroqui, L.; Quesada, I.; Gustafsson, J.A.; Nadal, A. Molecular mechanisms involved in the non-monotonic effect of bisphenol-A on Ca2+ entry in mouse pancreatic β-cells. Sci. Rep. 2017, 7, 11770. [Google Scholar] [CrossRef]
- Cano-Sancho, G.; Smith, A.; La Merrill, M.A. Triphenyl phosphate enhances adipogenic differentiation, glucose uptake and lipolysis via endocrine and noradrenergic mechanisms. Toxicol. Vitr. 2017, 40, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Graham, J.L.; Gonzalez, E.A.; La Frano, M.R.; Petropoulou, S.-S.E.; Park, J.-S.; Newman, J.W.; Stanhope, K.L.; Havel, P.J.; La Merrill, M.A. Perinatal triphenyl phosphate exposure accelerates type 2 diabetes onset and increases adipose accumulation in UCD-type 2 diabetes mellitus rats. Reprod. Toxicol. 2017, 68, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, E.A.; Patel, V.J.; Tillery, T.S.; Yasrebi, A.; Shen, J.; Guo, G.L.; Marco, S.M.; Buckley, B.T.; Roepke, T.A. Organophosphate flame-retardants alter adult mouse homeostasis and gene expression in a sex-dependent manner potentially through interactions with era. Toxicol. Sci. 2018, 162, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, S.; Yan, J.; Teng, M.; Meng, Z.; Li, R.; Zhou, Z.; Zhu, W. Effects of triphenyl phosphate exposure during fetal development on obesity and metabolic dysfunctions in adult mice: Impaired lipid metabolism and intestinal dysbiosis. Environ. Pollut. 2019, 246, 630–638. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Ha, C.-M.; Kim, S.-A.; Thoudam, T.; Yoon, Y.-R.; Kim, D.-J.; Kim, H.-C.; Moon, H.-B.; Park, S.; Lee, I.-K.; et al. Low-Dose Persistent Organic Pollutants Impair Insulin Secretory Function of Pancreatic β-Cells: Human and In Vitro Evidence. Diabetes 2017, 66, 2669–2680. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.B.; Dail, M.B.; Chambers, J.E. In vitro effect of DDE exposure on the regulation of B-TC-6 pancreatic beta cell insulin secretion: A potential role in beta cell dysfunction and type 2 diabetes mellitus. Toxicol. Mech. Methods 2021, 31, 667–673. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Pipeleers, D.G.; Ling, Z.; Welsh, N.; Hellerström, C.; Andersson, A. Major species differences between humans and rodents in the susceptibility to pancreatic beta-cell injury. Proc. Natl. Acad. Sci. USA 1994, 91, 9253–9256. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Mandrup-Poulsen, T. A choice of death - The signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Kutlu, B.; Cardozo, A.K.; Darville, M.I.; Kruhøffer, M.; Magnusson, N.; Ørntoft, T.; Eizirik, D.L. Discovery of Gene Networks Regulating Cytokine-Induced Dysfunction and Apoptosis in Insulin-Producing INS-1 Cells. Diabetes 2003, 52, 2701–2719. [Google Scholar] [CrossRef] [Green Version]
- Ortis, F.; Cardozo, A.K.; Crispim, D.; Störling, J.; Mandrup-Poulsen, T.; Eizirik, D.L. Cytokine-Induced Proapoptotic Gene Expression in Insulin-Producing Cells Is Related to Rapid, Sustained, and Nonoscillatory Nuclear Factor-κB Activation. Mol. Endocrinol. 2006, 20, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Santin, I.; Dos Santos, R.S.; Eizirik, D.L. Pancreatic beta cell survival and signaling pathways: Effects of type 1 diabetes-associated genetic variants. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1433, pp. 21–54. [Google Scholar]
- Nadal, A.; Soria, B. Glucose Metabolism Regulates Cytosolic Ca2+ in the Pancreatic β-Cell by Three Different Mechanisms. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 1997; Volume 426, pp. 235–243. [Google Scholar]
- Valdeolmillos, M.; Nadal, A.; Contreras, D.; Soria, B. The relationship between glucose-induced K+ATP channel closure and the rise in [Ca2+]i in single mouse pancreatic beta-cells. J. Physiol. 1992, 455, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.S.; Marroqui, L.; Velayos, T.; Olazagoitia-Garmendia, A.; Jauregi-Miguel, A.; Castellanos-Rubio, A.; Eizirik, D.L.; Castaño, L.; Santin, I. DEXI, a candidate gene for type 1 diabetes, modulates rat and human pancreatic beta cell inflammation via regulation of the type I IFN/STAT signalling pathway. Diabetologia 2019, 62, 459–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dos Santos, R.S.; Medina-Gali, R.M.; Babiloni-Chust, I.; Marroqui, L.; Nadal, A. In Vitro Assays to Identify Metabolism-Disrupting Chemicals with Diabetogenic Activity in a Human Pancreatic β-Cell Model. Int. J. Mol. Sci. 2022, 23, 5040. https://doi.org/10.3390/ijms23095040
Dos Santos RS, Medina-Gali RM, Babiloni-Chust I, Marroqui L, Nadal A. In Vitro Assays to Identify Metabolism-Disrupting Chemicals with Diabetogenic Activity in a Human Pancreatic β-Cell Model. International Journal of Molecular Sciences. 2022; 23(9):5040. https://doi.org/10.3390/ijms23095040
Chicago/Turabian StyleDos Santos, Reinaldo Sousa, Regla María Medina-Gali, Ignacio Babiloni-Chust, Laura Marroqui, and Angel Nadal. 2022. "In Vitro Assays to Identify Metabolism-Disrupting Chemicals with Diabetogenic Activity in a Human Pancreatic β-Cell Model" International Journal of Molecular Sciences 23, no. 9: 5040. https://doi.org/10.3390/ijms23095040