
Ubiquitin and Ubiquitin-like Proteins in Cancer, Neurodegenerative Disorders, and Heart Diseases


Abstract
:1. Introduction
2. Ub/Ubl Modifications in Disease Pathogenesis and Treatment
2.1. Ubiquitin-Proteasome System
2.1.1. Abnormal Ub Pathways in Cancer
2.1.2. UPS Inhibitors Used to Treat Cancer
2.1.3. Abnormal Ub Pathway in Neurodegenerative Diseases
2.1.4. UPS Inhibitors as Treatment for Neurodegenerative Diseases
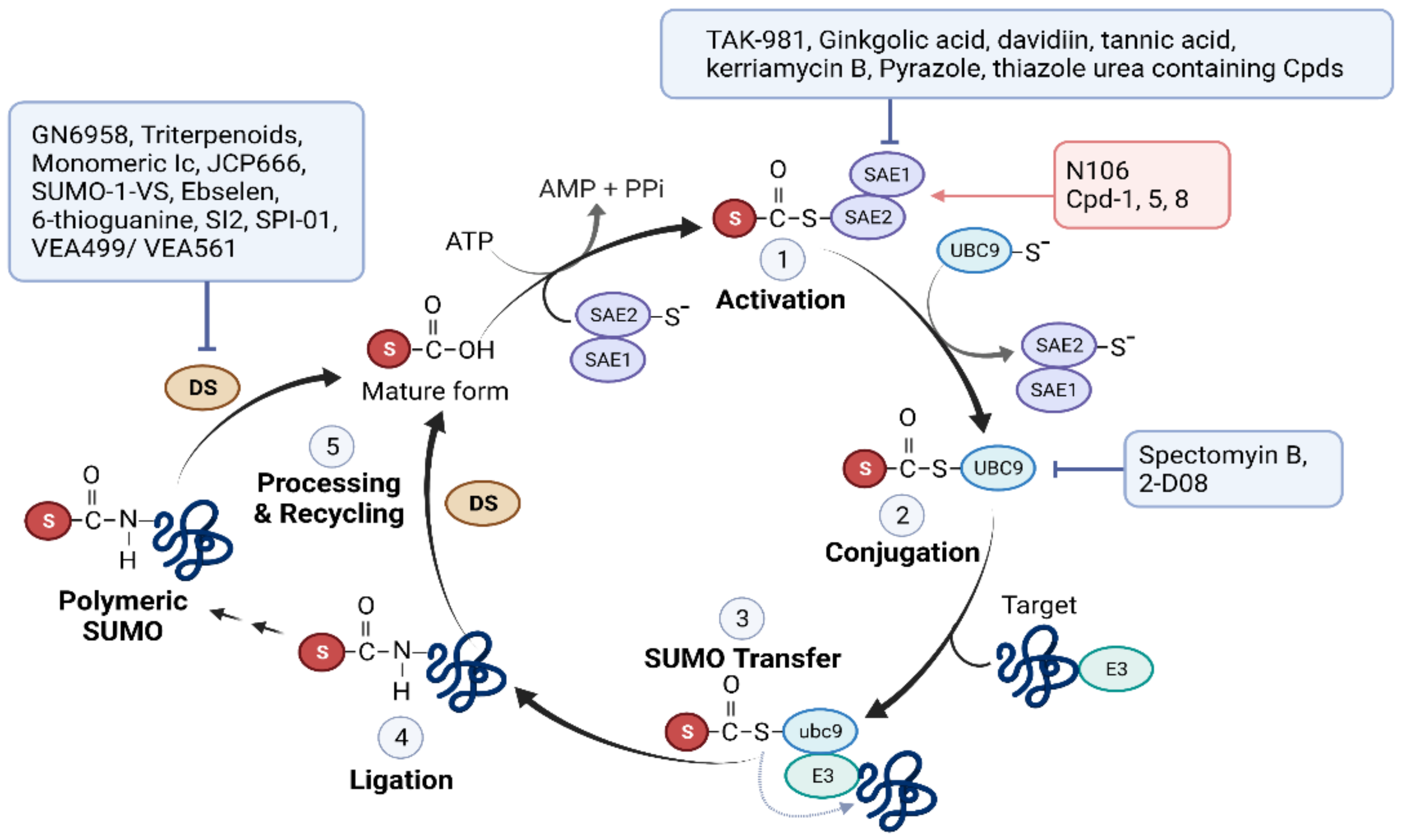
2.2. SUMOylation
2.2.1. Dysregulation of the SUMO Pathway in Cancer
2.2.2. SUMO Pathway Inhibitors Used to Treat Cancer

{kind=link}
{kind=link}
{kind=link}
| Compound ID | Description | Stage | |
|---|---|---|---|
| E1 | TAK-981 (Subasumstat) | The first-in-Class SAE Inhibitor | Phase I/II trials (https://clinicaltrials.gov/ct2/show/NCT03648372, accessed on 1 March 2022) |
| Ginkgolic acid, davidiin, tannic acid, kerriamycin B, Pyrazole, and thiazole urea containing Cpds | SAE inhibitor | Preclinical [93,94,95,96,112] | |
| E2 | 2-D08, Spectomyin B | UBC9 inhibitor | Preclinical [101,102,103,104,105,106] |
| SENP | GN6958, Triterpenoids, Monomeric Ic | SENP1 inhibitor | Preclinical [108,110,111] |
| JCP666 and its analogues | SENP1/2 inhibitor | Preclinical [113] | |
| SUMO-1-VS, Ebselen and 6-thioguanine | SENP2 inhibitor | Preclinical [114,115] | |
| SI2 | SENP1/2/3 inhibitor | Preclinical [109] | |
| SPI-01 | SENP1/2/7 inhibitor | Preclinical [109] | |
| VEA499/VEA561 | SENP1/2/6/7 inhibitor | Preclinical [113] |
2.2.3. Role of SUMOylation in Neurodegenerative Disease Pathogenesis
2.2.4. Therapeutics Targeting the SUMO Pathway in Neurodegenerative Diseases
2.2.5. Dysregulation of SUMOylation in Heart Disease
2.2.6. Therapeutics Targeting the SUMO Pathway
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef]
- Hochstrasser, M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat. Cell Biol. 2000, 2, E153–E157. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.W.; Ryu, K.Y. Cellular ubiquitin pool dynamics and homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Ruosaari, S.; Hienonen-Kempas, T.; Puustinen, A.; Sarhadi, V.K.; Hollmén, J.; Knuutila, S.; Saharinen, J.; Wikman, H.; Anttila, S. Pathways affected by asbestos exposure in normal and tumour tissue of lung cancer patients. BMC Med. Genom. 2008, 1, 55. [Google Scholar] [CrossRef] [Green Version]
- McHugh, A.; Fernandes, K.; South, A.P.; Mellerio, J.E.; Salas-Alanís, J.C.; Proby, C.M.; Leigh, I.M.; Saville, M.K. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: The identification of mechanisms of differential sensitivity. Oncotarget 2018, 9, 20265–20281. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.W.; Ali, M.; Wood, T.E.; Wong, D.; Maclean, N.; Wang, X.; Gronda, M.; Skrtic, M.; Li, X.; Hurren, R.; et al. The ubiquitin-activating enzyme E1 as a therapeutic target for the treatment of leukemia and multiple myeloma. Blood 2010, 115, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhao, J.; Yang, F.; Liu, H.; Qi, W. Ubiquitin conjugating enzyme E2 L3 promoted tumor growth of NSCLC through accelerating p27kip1 ubiquitination and degradation. Oncotarget 2017, 8, 84193–84203. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhao, H.; Yu, C.; Kang, Y.; Yang, X. Targeting Ubiquitin-Specific Protease 7 (USP7) in Cancer: A New Insight to Overcome Drug Resistance. Front. Pharmacol. 2021, 12, 648491. [Google Scholar] [CrossRef]
- Shen, J.D.; Fu, S.Z.; Ju, L.L.; Wang, Y.F.; Dai, F.; Liu, Z.X.; Ji, H.Z.; Shao, J.G.; Bian, Z.L. High expression of ubiquitin-conjugating enzyme E2A predicts poor prognosis in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 7362–7368. [Google Scholar] [CrossRef]
- Luo, H.; Qin, Y.; Reu, F.; Ye, S.; Dai, Y.; Huang, J.; Wang, F.; Zhang, D.; Pan, L.; Zhu, H.; et al. Microarray-based analysis and clinical validation identify ubiquitin-conjugating enzyme E2E1 (UBE2E1) as a prognostic factor in acute myeloid leukemia. J. Hematol. Oncol. 2016, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Gallo, L.H.; Ko, J.; Donoghue, D.J. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 2017, 16, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Hong, W.; Ruan, S.; Guan, R.; Tu, L.; Huang, B.; Hou, B.; Jian, Z.; Ma, L.; Jin, H. Genome-wide profiling of prognostic alternative splicing pattern in pancreatic cancer. Front. Oncol. 2019, 9, 773. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J.; Welcker, M.; Clurman, B.E. Tumor suppression by the Fbw7 ubiquitin ligase: Mechanisms and opportunities. Cancer Cell. 2014, 26, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Yu, N. Ubiquitin-specific protease 7 expression is a prognostic factor in epithelial ovarian cancer and correlates with lymph node metastasis. OncoTargets Ther. 2016, 9, 1559–1569. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Niu, C.; Yang, Y.; Wang, Y.; Lu, M. Expression of HAUSP in gliomas correlates with disease progression and survival of patients. Oncol. Rep. 2013, 29, 1730–1736. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, Y.; Yang, X.H.; Kang, T.; Zhao, Y.; Wang, C.; Evers, B.M.; Zhou, B.P. The Deubiquitinase USP28 Stabilizes LSD1 and Confers Stem-Cell-like Traits to Breast Cancer Cells. Cell Rep. 2013, 5, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Diefenbacher, M.E.; Popov, N.; Blake, S.M.; Schülein-Völk, C.; Nye, E.; Spencer-Dene, B.; Jaenicke, L.A.; Eilers, M.; Behrens, A. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J. Clin. Investig. 2014, 124, 3407–3418. [Google Scholar] [CrossRef] [Green Version]
- Hou, K.; Zhu, Z.; Wang, Y.; Zhang, C.; Yu, S.; Zhu, Q.; Yan, B. Overexpression and Biological Function of Ubiquitin-Specific Protease 42 in Gastric Cancer. PLoS ONE 2016, 11, e0152997. [Google Scholar] [CrossRef]
- Shah, J.; Usmani, S.; Stadtmauer, E.A.; Rifkin, R.M.; Berenson, J.R.; Berdeja, J.G.; Lyons, R.M.; Klippel, Z.; Chang, Y.L.; Niesvizky, R. Oprozomib, pomalidomide, and Dexamethasone in Patients with Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 570–578.e1. [Google Scholar] [CrossRef]
- Gallerani, E.; Zucchetti, M.; Brunelli, D.; Marangon, E.; Noberasco, C.; Hess, D.; Delmonte, A.; Martinelli, G.; Böhm, S.; Driessen, C.; et al. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur. J. Cancer 2013, 49, 290–296. [Google Scholar] [CrossRef]
- Vogl, D.T.; Martin, T.G.; Vij, R.; Hari, P.; Mikhael, J.R.; Siegel, D.; Wu, K.L.; Delforge, M.; Gasparetto, C. Phase I/II study of the novel proteasome inhibitor delanzomib (CEP-18770) for relapsed and refractory multiple myeloma. Leuk. Lymphoma 2017, 58, 1872–1879. [Google Scholar] [CrossRef]
- Nunes, A.T.; Annunziata, C.M. Proteasome inhibitors: Structure and function. Semin. Oncol. 2017, 6, 377–380. [Google Scholar] [CrossRef]
- Barghout, S.H.; Schimmer, A.D. E1 Enzymes as Therapeutic Targets in Cancer. Pharmacol. Rev. 2021, 73, 1–58. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Antao, A.M.; Tyagi, A.; Kim, K.S.; Ramakrishna, S. Advances in Deubiquitinating Enzyme Inhibition and Applications in Cancer Therapeutics. Cancers 2020, 12, 1579. [Google Scholar] [CrossRef] [PubMed]
- Rowinsky, E.K.; Paner, A.; Berdeja, J.G.; Paba-Prada, C.; Venugopal, P.; Porkka, K.; Gullbo, J.; Linder, S.; Loskog, A.; Richardson, P.G.; et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Investig. New Drugs 2020, 38, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Katsnelson, A. Next-generation proteasome inhibitor approved in multiple myeloma. Nat. Biotechnol. 2012, 30, 1011–1102. [Google Scholar] [CrossRef]
- Shirley, M. Ixazomib: First Global Approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Rotblat, B.; Southwell, A.L.; Ehrnhoefer, D.E.; Skotte, N.H.; Metzler, M.; Franciosi, S.; Leprivier, G.; Somasekharan, S.P.; Barokas, A.; Deng, Y.; et al. HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. USA 2014, 111, 3032–3037. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef]
- Kwak, Y.-D.; Wang, B.; Li, J.J.; Wang, R.; Deng, Q.; Diao, S.; Chen, Y.; Xu, R.; Masliah, E.; Xu, H.; et al. Upregulation of the E3 ligase NEDD4-1 by Oxidative Stress Degrades IGF-1 Receptor Protein in Neurodegeneration. J. Neurosci. 2012, 32, 10971–10981. [Google Scholar] [CrossRef]
- Liu, Q.Y.; Lei, J.X.; Sikorska, M.; Liu, R. A novel brain-enriched E3 ubiquitin ligase RNF182 is up regulated in the brains of Alzheimer’s patients and targets ATP6V0C for degradation. Mol. Neurodegener. 2008, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.Y.; Park, H.R.; Lee, S.J.; Lee, S.H.; Kim, J.S.; Jung, Y.S.; Hwang, S.H.; Ha, N.C.; Seol, W.G.; Lee, J.; et al. Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab. Investig. 2013, 93, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Zucchelli, S.; Marcuzzi, F.; Codrich, M.; Agostoni, E.; Vilotti, S.; Biagioli, M.; Pinto, M.; Carnemolla, A.; Santoro, C.; Gustincich, S.; et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation. J. Biol. Chem. 2011, 286, 25108–25117. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hebron, M.; Shi, W.; Lonskaya, I.; Moussa, C.E. Ubiquitin specific protease-13 independently regulates parkin ubiquitination and alpha-synuclein clearance in alpha-synucleinopathies. Hum. Mol. Genet. 2019, 28, 548–560. [Google Scholar] [CrossRef]
- Van Leeuwen, F.W.; de Kleijn, D.P.; van den Hurk, H.H.; Neubauer, A.; Sonnemans, M.A.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.; Salehi, A.; Martens, G.J.; et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down’s patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef]
- Krutauz, D.; Reis, N.; Nakasone, M.A.; Siman, P.; Zhang, D.; Kirkpatrick, D.S.; Gygi, S.P.; Brik, A.; Fushman, D.; Glickman, M.H. Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 2014, 10, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Sun, X.; Fy, H.; Hw, O.; Hilgenberg, L.G.W.; Hol, E.M.; van Leeuwen, F.W.; Smith, M.A.; O’Dowd, D.K.; Schreiber, S.S. Mutant ubiquitin found in Alzheimer’s disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 2007, 14, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Chen, J.; Cao, W.; Yang, L.; Chen, Q.; He, J.; Yi, Q.; Huang, H.; Zhang, E.; Cai, Z. The many substrates and functions of NEDD4-1. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Rodrigues, E.M.; Scudder, S.L.; Goo, M.S.; Patrick, G.N. Aβ-Induced Synaptic Alterations Require the E3 Ubiquitin Ligase Nedd4-1. J. Neurosci. 2016, 36, 1590–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, T.L.; Rissman, R.A.; Mishizen-Eberz, A.J.; Wolfe, B.B.; Hamilton, R.L.; Gandy, S.; Armstrong, D.M. Differential preservation of AMPA receptor subunits in the hippocampi of Alzheimer’s disease patients according to Braak stage. Exp. Neurol. 2004, 187, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Pasini, S.; Corona, C.; Robador, P.; Song, C.; Patel, H.; Lefort, R. Dysfunction of the ubiquitin ligase E3A Ube3A/E6-AP contributes to synaptic pathology in Alzheimer’s disease. Commun. Biol. 2019, 2, 111. [Google Scholar] [CrossRef] [PubMed]
- Maranga, C.; Fernandes, T.G.; Bekman, E.; Da Rocha, S.T. Angelman syndrome: A journey through the brain. FEBS J. 2020, 287, 2154–2175. [Google Scholar] [CrossRef]
- Singh, B.K.; Vatsa, N.; Kumar, V.; Shekhar, S.; Sharma, A.; Jana, N.R. Ube3a deficiency inhibits amyloid plaque formation in APPswe/PS1δE9 mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4042–4054. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of α-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. 2010, 25, S32–S39. [Google Scholar] [CrossRef]
- Clark, E.H.; Vázquez de la Torre, A.; Hoshikawa, T.; Briston, T. Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 2021, 296, 100209. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Setsuie, R.; Wang, Y.L.; Mochizuki, H.; Osaka, H.; Hayakawa, H.; Ichihara, N.; Li, H.; Furuta, A.; Sano, Y.; Sun, Y.J.; et al. Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem. Int. 2007, 50, 119–129. [Google Scholar] [CrossRef]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef] [Green Version]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.E.; Hallett, P.J.; Moens, T.; Smith, G.; Mangano, E.; Kim, H.T.; Goldberg, A.L.; Liu, J.L.; Isacson, O.; Tofaris, G.K. Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol. Dis. 2014, 64, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Boselli, M.; Lee, B.H.; Robert, J.; Prado, M.A.; Min, S.W.; Cheng, C.; Silva, M.C.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An Inhibitor of the Proteasomal Deubiquitinating Enzyme USP14 Induces Tau Elimination in Cultured Neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, J.; von Stockum, S.; Marchesan, E.; Caicci, F.; Ferrari, V.; Rakovic, A.; Klein, C.; Antonini, A.; Bubacco, L.; Ziviani, E. USP14 inhibition corrects an in vivo model of impaired mitophagy. EMBO Mol. Med. 2018, 10, e9014. [Google Scholar] [CrossRef]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.J.; Lee, M.J. Dual Function of USP14 Deubiquitinase in Cellular Proteasomal Activity and Autophagic Flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Kemp, M.; Stockley, M.; Jones, A. Cyanopyrrolidines as Dub Inhibitors for the Treatment of Cancer. WO 2017009650 (A1) 19 January 2017. [Google Scholar]
- Pukass, K.; Richter-Landsberg, C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents alpha-synuclein aggregate formation by activating the autophagic pathway: Implications for multiple system atrophy. Front. Cell. Neurosci. 2015, 9, 163. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.J.; Lee, D.; Xu, W.J.; Cho, E.; Choi, S.; Min, S.; Park, S.; Chung, J. Loss of UCHL1 rescues the defects related to Parkinson’s disease by suppressing glycolysis. Sci. Adv. 2021, 7, eabg4574. [Google Scholar] [CrossRef]
- Kluge, A.F.; Lagu, B.R.; Maiti, P.; Jaleel, M.; Webb, M.; Malhotra, J.; Mallat, A.; Srinivas, P.A.; Thompson, J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018, 28, 2655–2659. [Google Scholar] [CrossRef]
- Cartier, A.E.; Djakovic, S.N.; Salehi, A.; Wilson, S.M.; Masliah, E.; Patrick, G.N. Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci. 2009, 29, 7857–7868. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.; Bedekovics, T.; Chesi, M.; Bergsagel, P.L.; Galardy, P.J. UCHL1 is a biomarker of aggressive multiple myeloma required for disease progression. Oncotarget 2015, 6, 40704–40718. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 2011, 18, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Zhongxia, Y.; Hui, S.; Mutian, J.; Jintao, Z.; Wenwen, W.; Qi, L.; Lining, Z.; Wei, Z. USP1–UAF1 deubiquitinase complex stabilizes TBK1 and enhances antiviral responses. J. Exp. Med. 2017, 214, 3553–3563. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Patten, S.A.; Aggad, D.; Martinez, J.; Tremblay, E.; Petrillo, J.; Armstrong, G.A.; La Fontaine, A.; Maios, C.; Liao, M.; Ciura, S.; et al. Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2017, 2, e97152. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, Y.; Pang, Z.; Guo, F.; Qin, Q.; Yin, T.; Sang, Y.; Feng, C.; Li, X.; Jiang, L.; et al. Knockdown of SUMO-activating enzyme subunit 2 (SAE2) suppresses cancer malignancy and enhances chemotherapy sensitivity in small cell lung cancer. J. Hematol. Oncol. 2015, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dünnebier, T.; Bermejo, J.L.; Haas, S.; Fischer, H.P.; Pierl, C.B.; Justenhoven, C.; Brauch, H.; Baisch, C.; Gilbert, M.; Harth, V.; et al. Polymorphisms in the UBC9 and PIAS3 genes of the SUMO-conjugating system and breast cancer risk. Breast Cancer Res. Treat. 2010, 121, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D., Jr.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef] [Green Version]
- Tokarz, P.; Woźniak, K. SENP Proteases as Potential Targets for Cancer Therapy. Cancers 2021, 13, 2059. [Google Scholar] [CrossRef]
- Stefanska, B.; Cheishvili, D.; Suderman, M.; Arakelian, A.; Huang, J.; Hallett, M.; Han, Z.G.; Al-Mahtab, M.; Akbar, S.M.; Khan, W.A.; et al. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets. Clin. Cancer Res. 2014, 20, 3118–3132. [Google Scholar] [CrossRef] [Green Version]
- Tuccilli, C.; Baldini, E.; Sorrenti, S.; Di Gioia, C.; Bosco, D.; Ascoli, V.; Mian, C.; Barollo, S.; Rendina, R.; Coccaro, C.; et al. Papillary Thyroid Cancer Is Characterized by Altered Expression of Genes Involved in The Sumoylation Process. J. Biol. Regul. Homeost. Agents 2015, 29, 655–662. [Google Scholar]
- Moschos, S.J.; Smith, A.P.; Mandic, M.; Athanassiou, C.; Watson-Hurst, K.; Jukic, D.M.; Edington, H.D.; Kirkwood, J.M.; Becker, D. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene 2007, 26, 4216–4225. [Google Scholar] [CrossRef] [Green Version]
- Hoefer, J.; Schäfer, G.; Klocker, H.; Erb, H.H.; Mills, I.G.; Hengst, L.; Puhr, M.; Culig, Z. PIAS1 is increased in human prostate cancer and enhances proliferation through inhibition of p21. Am. J. Pathol. 2012, 180, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Vecchione, L.; Gambino, V.; Raaijmakers, J.; Schlicker, A.; Fumagalli, A.; Russo, M.; Villanueva, A.; Beerling, E.; Bartolini, A.; Mollevi, D.G.; et al. A Vulnerability of a Subset of Colon Cancers with Potential Clinical Utility. Cell 2016, 165, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Bawa-Khalfe, T.; Lu, L.S.; Zuo, Y.; Huang, C.; Dere, R.; Lin, F.M.; Yeh, E.T. Differential expression of SUMO-specific protease 7 variants regulates epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2012, 109, 17466–17471. [Google Scholar] [CrossRef] [Green Version]
- Brackett, C.M.; Blagg, B.S.J. Current Status of SUMOylation Inhibitors. Curr. Med. Chem. 2021, 28, 3892–3912. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef]
- Takemoto, M.; Kawamural, Y.; Hirohama, M.; Yamaguchi, Y.; Handa, H.; Saitoh, H.; Nakao, Y.; Kawada, M.; Khalid, K.; Koshino, H.; et al. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. J. Antibiot. 2014, 67, 335–338. [Google Scholar] [CrossRef]
- Suzawa, M.; Miranda, D.A.; Ramos, K.A.; Ang, K.K.; Faivre, E.J.; Wilson, C.G.; Caboni, L.; Arkin, M.R.; Kim, Y.S.; Fletterick, R.J.; et al. A gene-expression screen identifies a non-toxic sumoylation inhibitor that mimics SUMO-less human LRH-1 in liver. eLife 2015, 4, e09003. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Uramoto, M.; Saitoh, H.; Kawasaki, H.; Osada, H.; Yoshida, M. Kerriamycin B inhibits protein SUMOylation. J. Antibiot. 2009, 62, 221–224. [Google Scholar] [CrossRef]
- He, X.; Riceberg, J.; Soucy, T.; Koenig, E.; Minissale, J.; Gallery, M.; Bernard, H.; Yang, X.; Liao, H.; Rabino, C.; et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 2017, 13, 1164–1171. [Google Scholar] [CrossRef]
- Langston, S.P.; Grossman, S.; England, D.; Afroze, R.; Bence, N.; Bowman, D.; Bump, N.; Chau, R.; Chuang, B.C.; Claiborne, C.; et al. Discovery of TAK-981, a First-in-Class Inhibitor of SUMO-Activating Enzyme for the Treatment of Cancer. J. Med. Chem. 2021, 64, 2501–2520. [Google Scholar] [CrossRef] [PubMed]
- Lightcap, E.S.; Yu, P.; Grossman, S.; Song, K.; Khattar, M.; Xega, K.; He, X.; Gavin, J.M.; Imaichi, H.; Garnsey, J.J.; et al. A small-molecule SUMOylation inhibitor activates antitumor immune responses and potentiates immune therapies in preclinical models. Sci. Transl. Med. 2021, 13, eaba7791. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Kim, Y.S.; Keyser, S.G.; Schneekloth, J.S., Jr. Synthesis of 2′,3′,4′-trihydroxyflavone (2-D08), an inhibitor of protein sumoylation. Bioorg. Med. Chem. Lett. 2014, 24, 1094–1097. [Google Scholar] [CrossRef] [Green Version]
- Hirohama, M.; Kumar, A.; Fukuda, I.; Matsuoka, S.; Igarashi, Y.; Saitoh, H.; Takagi, M.; Shin-ya, K.; Honda, K.; Kondoh, Y.; et al. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem. Biol. 2013, 8, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Philips, M.R.; Chen, Y.; Lu, L.; Dai, W. K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J. Biol. Chem. 2018, 293, 17574–17581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Chen, X.; Li, M.; Tan, J.; Zhang, Y.; Yuan, W.; Zhou, J.; Wang, G. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019, 513, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zheng, L.; Hu, K.; Wang, X.; Zhang, R.; Zou, Y.; Zhong, L.; Wang, S.; Wu, Y.; Kang, T. SUMOylation stabilizes hSSB1 and enhances the recruitment of NBS1 to DNA damage sites. Signal Transduct. Target. Ther. 2020, 5, 80. [Google Scholar] [CrossRef]
- Baik, H.; Boulanger, M.; Hosseini, M.; Kowalczyk, J.; Zaghdoudi, S.; Salem, T.; Sarry, J.E.; Hicheri, Y.; Cartron, G.; Piechaczyk, M.; et al. Targeting the SUMO pathway primes all-trans retinoic acid-induced differentiation of non-promyelocytic acute myeloid leukemias. Cancer Res. 2018, 78, 2601–2613. [Google Scholar] [CrossRef] [Green Version]
- Zlotkowski, K.; Hewitt, W.M.; Sinniah, R.S.; Tropea, J.E.; Needle, D.; Lountos, G.T.; Barchi, J.J., Jr.; Waugh, D.S.; Schneekloth, J.S., Jr. A Small-Molecule Microarray Approach for the Identification of E2 Enzyme Inhibitors in Ubiquitin-Like Conjugation Pathways. SLAS Discov. Adv. Life Sci. 2017, 22, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Uno, M.; Koma, Y.; Ban, H.S.; Nakamura, H. Discovery of 1-[4-(N-benzylamino)phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP)1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5169–5173. [Google Scholar] [CrossRef]
- Wen, D.; Xu, Z.; Xia, L.; Liu, X.; Tu, Y.; Lei, H.; Wang, W.; Wang, T.; Song, L.; Ma, C.; et al. Important role of SUMOylation of Spliceosome factors in prostate cancer cells. J. Proteome Res. 2014, 13, 3571–3582. [Google Scholar] [CrossRef]
- Huang, W.; He, T.; Chai, C.; Yang, Y.; Zheng, Y.; Zhou, P.; Qiao, X.; Zhang, B.; Liu, Z.; Wang, J.; et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS ONE 2012, 7, e37693. [Google Scholar] [CrossRef]
- Wu, J.; Lei, H.; Zhang, J.; Chen, X.; Tang, C.; Wang, W.; Xu, H.; Xiao, W.; Gu, W.; Wu, Y. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget 2016, 7, 58995–59005. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ito, A.; Hirohama, M.; Yoshida, M.; Zhang, K.Y. Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg. Med. Chem. Lett. 2016, 26, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Albrow, V.E.; Ponder, E.L.; Fasci, D.; Békés, M.; Deu, E.; Salvesen, G.S.; Bogyo, M. Development of small molecule inhibitors and probes of human SUMO deconjugating proteases. Chem. Biol. 2011, 18, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Hemelaar, J.; Borodovsky, A.; Kessler, B.M.; Reverter, D.; Cook, J.; Kolli, N.; Gan-Erdene, T.; Wilkinson, K.D.; Gill, G.; Lima, C.D.; et al. Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins. Mol. Cell. Biol. 2014, 24, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Bernstock, J.D.; Ye, D.; Smith, J.A.; Lee, Y.J.; Gessler, F.A.; Yasgar, A.; Kouznetsova, J.; Jadhav, A.; Wang, Z.; Pluchino, S.; et al. Quantitative high-throughput screening identifies cytoprotective molecules that enhance SUMO conjugation via the inhibition of SUMO-specific protease (SENP)2. FASEB J. 2018, 32, 1677–1691. [Google Scholar] [CrossRef] [Green Version]
- Yau, T.Y.; Molina, O.; Courey, A.J. SUMOylation in development and neurodegeneration. Development 2020, 147, dev175703. [Google Scholar] [CrossRef]
- Yang, W.; Sheng, H.; Warner, D.S.; Paschen, W. Transient global cerebral ischemia induces a massive increase in protein sumoylation. J. Cereb. Blood Flow Metab. 2008, 28, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Bernstock, J.D.; Ye, D.G.; Griffin, A.; Lee, Y.-J.; Lynch, J.; Latour, L.L.; Friedman, G.K.; Maric, D.; Hallenbeck, J.M. Cerebral ischemia increases small ubiquitin-like modifier conjugation within human penumbral tissue: Radiological-pathological correlation. Front. Neurol. 2018, 8, 738. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Castri, P.; Bembry, J.; Maric, D.; Auh, S.; Hallenbeck, J.M. SUMOylation participates in induction of ischemic tolerance. J. Neurochem. 2009, 109, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.; Song, J.H.; Kim, D.K.; Park, M.H.; Jo, S.A.; Koh, Y.H. Ubc9 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population: A genetic association study. Neurosci. Lett. 2009, 465, 272–275. [Google Scholar] [CrossRef]
- Weeraratna, A.T.; Kalehua, A.; Deleon, I.; Bertak, D.; Maher, G.; Wade, M.S.; Lustig, A.; Becker, K.G.; Wood, W., 3rd; Walker, D.G.; et al. Alterations in immunological and neurological gene expression patterns in Alzheimer’s disease tissues. Exp. Cell Res. 2007, 313, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Dale, E.; Staniszewski, A.; Zhang, H.; Saeed, F.; Sakurai, M.; Fa, M.; Orozco, I.; Michelassi, F.; Akpan, N.; et al. Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer’s disease. Sci. Rep. 2014, 4, 7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Q.; Sarge, K.D. Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun. 2008, 374, 673–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.B.; Xia, Y.Y.; Shu, X.J.; Liu, Z.C.; Feng, Y.; Liu, X.H.; Yu, G.; Yin, G.; Xiong, Y.S.; Zeng, K.; et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baulac, S.; Lu, H.; Strahle, J.; Yang, T.; Goldberg, M.S.; Shen, J.; Schlossmacher, M.G.; Lemere, C.A.; Lu, Q.; Xia, W. Increased DJ-1 expression under oxidative stress and in Alzheimer’s disease brains. Mol. Neurodegener. 2009, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, H.D. Mutual interactions between the SUMO and ubiquitin systems: A plea of no contest. Trends Cell Biol. 2005, 15, 525–532. [Google Scholar] [CrossRef]
- Ramírez-Jarquín, U.N.; Sharma, M.; Zhou, W.; Shahani, N.; Subramaniam, S. Deletion of SUMO1 attenuates behavioral and anatomical deficits by regulating autophagic activities in Huntington disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2107187119. [Google Scholar] [CrossRef]
- Marsh, D.T.; Das, S.; Ridell, J.; Smid, S.D. Structure-activity relationships for flavone interactions with amyloid beta reveal a novel anti-aggregatory and neuroprotective effect of 2′,3′,4′-trihydroxyflavone (2-D08). Bioorg. Med. Chem. 2017, 25, 3827–3834. [Google Scholar] [CrossRef]
- Krajnak, K.; Dahl, R. Small molecule SUMOylation activators are novel neuroprotective agents. Bioorg. Med. Chem. Lett. 2018, 28, 405–409. [Google Scholar] [CrossRef]
- Yang, W.; Sheng, H.; Homi, H.M.; Warner, D.S.; Paschen, W. Cerebral ischemia/stroke and small ubiquitin-like modifier (SUMO) conjugation—A new target for therapeutic intervention? J. Neurochem. 2008, 106, 989–999. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Peruzzotti-Jametti, L.; Leonardi, T.; Vicario, N.; Ye, D.; Lee, Y.J.; Maric, D.; Johnson, K.R.; Mou, Y.; Van Den Bosch, A.; et al. SUMOylation promotes survival and integration of neural stem cell grafts in ischemic stroke. EBioMedicine 2019, 42, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, L.; Wen, S.; Zhu, H.; Yu, W.; Moskowitz, I.P.; Shaw, G.M.; Finnell, R.H.; Schwartz, R.J. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Chen, L.; Ma, Y.; Yu, W.; Chang, J.; Moskowitz, I.P.; Wang, J. Enhanced desumoylation in murine hearts by overexpressed SENP2 leads to congenital heart defects and cardiac dysfunction. J. Mol. Cell. Cardiol. 2012, 52, 638–649. [Google Scholar] [CrossRef] [Green Version]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Kim, E.Y.; Zhang, Y.; Ye, B.; Segura, A.M.; Beketaev, I.; Xi, Y.T.; Yu, W.; Chang, J.; Li, F.Q.; Wang, J. Involvement of activated SUMO-2 conjugation in cardiomyopathy. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Zhang, Y.; Beketaev, I.; Segura, A.M.; Yu, W.; Xi, Y.T.; Chang, J.; Wang, J. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 78, 154–164. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, X.; Rong, J. SUMOylation as a Therapeutic Target for Myocardial Infarction. Front. Cardiovasc. Med. 2021, 8, 701583. [Google Scholar] [CrossRef]
- Du, C.; Chen, X.; Su, Q.; Lu, W.; Wang, Q.; Yuan, H.; Zhang, Z.; Wang, X.; Wu, H.; Qi, Y. The Function of SUMOylation and Its Critical Roles in Cardiovascular Diseases and Potential Clinical Implications. Int. J. Mol. Sci. 2021, 22, 10618. [Google Scholar] [CrossRef]
- Gupta, M.K.; Gulick, J.; Liu, R.; Wang, X.; Molkentin, J.D.; Robbins, J. Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ. Res. 2014, 115, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Chen, X.H.; Jiang, R.C.; Chen, S.Y.; Chen, K.F.; Zhu, X.; Zhang, X.L.; Huang, J.J.; Qin, Y.; Zhang, G.P.; et al. Ubc9 Attenuates Myocardial Ischemic Injury Through Accelerating Autophagic Flux. Front. Pharmacol. 2020, 11, 561306. [Google Scholar] [CrossRef]
- Gu, J.; Fan, Y.; Liu, X.; Zhou, L.; Cheng, J.; Cai, R.; Xue, S. SENP1 Protects against Myocardial Ischaemia/reperfusion Injury via a HIF1α-dependent Pathway. Cardiovasc. Res. 2014, 104, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Cai, R.; Gu, J.; Sun, H.; Liu, X.; Mei, W.; Qi, Y.; Xue, S.; Ren, S.; Rabinowitz, J.E.; Wang, Y.; et al. Induction of SENP1 in myocardium contributes to abnormities of mitochondria and cardiomyopathy. Mol. Cell. Cardiol. 2015, 79, 115–122. [Google Scholar] [CrossRef]
- Del Monte, F.; Harding, S.E.; Schmidt, U.; Matsui, T.; Kang, Z.B.; Dec, G.W.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999, 100, 2308–2311. [Google Scholar] [CrossRef] [Green Version]
- Tilemann, L.; Lee, A.; Ishikawa, K.; Aguero, J.; Rapti, K.; Santos-Gallego, C.; Kohlbrenner, E.; Fish, K.M.; Kho, C.; Hajjar, R.J. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013, 5, 211ra159. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef]
- Hu, W.; Xu, T.; Wu, P.; Pan, D.; Chen, J.; Chen, J.; Zhang, B.; Zhu, H.; Li, D. Luteolin improves cardiac dysfunction in heart failure rats by regulating sarcoplasmic reticulum Ca2+-ATPase 2a. Sci. Rep. 2017, 7, 41017. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; McLendon, P.M.; Gulick, J.; James, J.; Khalili, K.; Robbins, J. UBC9-mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ. Res. 2016, 118, 1894–1905. [Google Scholar] [CrossRef] [Green Version]
- Liebelt, F.; Vertegaal, A.C. Ubiquitin-dependent and independent roles of SUMO in proteostasis. Am. J. Physiol. Cell Physiol. 2016, 311, C284–C296. [Google Scholar] [CrossRef] [PubMed] [Green Version]


| Gene Name | Function in Cancer | Deregulation | Cancer Type | |
|---|---|---|---|---|
| E1 | UBA1 | Oncogene | ↑ | LNC [7], SCC [8], MM [9], PM in LC, TC [TCGA] |
| E2 | UBE2A | Oncogene | Mutation, ↑ | HCC [12] CML [13], PM in RCC, CC, HNC [TCGA] |
| UBE2C | Proto-oncogene | ↑ | BC, PC, CRC, OC, Lymphoma [14], PM in RCC, LC, PC [TCGA] | |
| UBE2D | Oncogene | ↑ | PM in RCC [TCGA] | |
| UBE2L | Oncogene | ↑ | NSCLC [10], PM in BC, OC [TCGA] | |
| UBE2N | Oncogene | ↑ | BC, PC, CRC, OC, Lymphoma [14], PM in LC [TCGA] | |
| UBE2S | Oncogene | ↑ | PM in RCC, LC, EC [TCGA] | |
| UBE2T | Oncogene | ↑ | PM in RCC, LC, OC [TCGA] | |
| E3 | APC3 (CDC27) | Tumor suppressor | Mutation, ↑ | PC [15], PM in RCC, CRC, LC, LNC [TCGA] |
| BRCA1 | Tumor suppressor | Mutation | Familial BC, OC [OMIM 113705] | |
| CBL | Proto-oncogene | Mutation | Leukemia [OMIM 165360] | |
| FBW7 | Tumor suppressor | Mutation | BRC, CRC, EC [16], PM in RCC [TCGA] | |
| MDM2 | Oncogene | ↑ | PM in EC, CC [TCGA] | |
| SKP2 | Oncogene | ↑ | PM in RCC, Melanoma, OC [TCGA] | |
| PAKN2 | Tumor suppressor | Mutation | LNC, OC [OMIM 602544] | |
| VHL | Tumor suppressor | Mutation | RCC [OMIM 608537], PM in LC, BC, SC [TCGA] | |
| DUB | BAP1 | Tumor suppressor | Mutation | BC, LC, RCC [OMIM 603089], PM in RCC [TCGA] |
| CYLD | Tumor suppressor/Oncogene | Mutation | Familial cylindromatosis, Trichoepithelioma [OMIM 605018] | |
| FANCL | Tumor suppressor | Mutation | Fanconi leukemia [OMIM 608111], PM in RCC, LC, UC [TCGA] | |
| TNFAIP3 | Tumor suppressor/Oncogene | Mutation, ↑ | BC, Lymphomas [OMIM 191163], PM in RCC [TCGA] | |
| USP4 | Oncogene | Mutation, ↑ | LNC [OMIM 603486], PM in RCC, LNC [TCGA] | |
| USP7 | Tumor suppressor/Oncogene | ↑ | OC [17], Glioma [18], PM in RCC [TCGA] | |
| USP14 | Oncogene | ↑ | PM in LC, HNC, OC [TCGA] | |
| USP28 | Oncogene | ↑ | BC [19], CRC [20] | |
| USP42 | Oncogene | ↑ | GC [21] |
| Gene Name | Deregulation Type | Disease | |
|---|---|---|---|
| Ub precursor | UBB | Missreading, misframed mutations | AD [OMIM 191339] |
| E3 | CHIP | ↑ | AD [38] |
| FBXO7 | Loss-of-function mutations | PD [OMIM 605648] | |
| HACE1 | ↓ | HD [39] | |
| HRD1 | ↓ | AD [40] | |
| LRSAM1 | Loss-of-function mutations | PD [OMIM 610933] | |
| NEDD4-1 | ↑ | AD, PD, ALS [41] | |
| PRKN (PARK2) | Loss-of-function mutations | PD [OMIM 602544] | |
| RNF182 | ↑ | AD [42] | |
| TRAF6 | ↑ | PD [43], HD [44] | |
| UBE3A | Loss-of-function mutations | AS [OMIM 601623] | |
| DUB | UCHL1 (PARK5) | ↓, Loss-of-function mutations | AD, PD [OMIM 191342] |
| USP13 | ↑ | PD [45] |
| Compound ID | Description | Stage |
|---|---|---|
| Pimozide | USP1 inhibitor | Phase II trials (https://clinicaltrials.gov/ct2/show/NCT03272503, accessed on 1 March 2022) |
| IU1, IU1 analogs and derivatives | USP14 inhibitor | Preclinical [64,65,66,67] |
| Cyanopyrrolidine derivatives, LDN57444 | UCHL1 inhibitor | Preclinical [68,69,70] |
| MTX652, MTX114, MF0094 | USP30 inhibitors | Preclinical [71] |
| Gene Name | Deregulation | Caner Type | |
|---|---|---|---|
| Modifier | SUMO1 | ↑ | PM in OC [TCGA] |
| SUMO2 | ↑ | PM in RC, EC, HCC [TCGA] | |
| SUMO3 | ↑ | PM in EC [TCGA] | |
| SUMO E1 | SAE1 | ↑ | PTC [87] PM in HCC, RC, TC [TCGA] |
| SAE2 | ↑ | BC [80], SCLC [81], PM in HCC, RC [TCGA] | |
| SUMO E2 | UBC9 | ↑ | Melanoma [88], PM in TC, HCC [TCGA] |
| SUMO E3 | PIAS1 | ↑ | PTC [87], PCA [89] |
| PIAS2 | ↓ | PTC [87], PM in TC [TCGA] | |
| PIAS3 | ↑ or ↓, Mutation | PM in RC, HCC [TCGA] | |
| PIAS4 | ↑ | PM in EC, PAC [TCGA] | |
| RANBP2 | ↑, Mutation | CRC [90] | |
| deSUMOylase | SENP1 | ↑ | PM in RC, HCC [TCGA] |
| SENP2 | ↓ or ↑, Mutation | PM in EC [TCGA] | |
| SENP3 | ↑ | PM in PAC [TCGA] | |
| SENP5 | ↑ | PM in RC, EC, HCC [TCGA] | |
| SENP6 | ↑ | PM in RC, TC [TCGA] | |
| SENP7 | Long↑; Short varient↓ | BC [91] | |
| ↑ | PM for HNC [TCGA] |
| Substrate | Substrate’s Function | Functional Impact | Disease |
|---|---|---|---|
| APP | Aβ generation | Negative regulation of Aβ aggregates levels | AD [123] |
| Tau | Microtubule stabilization | Induction of tau hyper-phosphorylation & inhibition of tau degradation | AD [124] |
| HTT | Microtubule-mediated transport and vesicle function | Increased cytotoxicity by specifically stabilizing mutant HTT via Rhes | HD [125] |
| α-Synuclein | PD pathogenesis | Maintanance of α-synuclein in a soluble form | PD [126] |
| DJ-1 | Anti-oxidative stress and transcriptional regulation | Essential for DJ-1 solubility and its function | PD [127] |
| Parkin | E3 Ub ligase | Induction of Parkin’s self-ubiquitination & nuclear translocation | PD [128] |
| Component | Expression | Regulation Pathway | Disease | |
|---|---|---|---|---|
| Modifier | SUMO1 | ↓ | Heart development, cardiac pathology | CHD, HF [134,136] |
| SUMO2/3 conjugates | ↑ | Cardiac pathology | HF [137] | |
| E2 | UBC9 | ↑ | Authophagy | MI, CM [141,142] |
| Deconjugase | SENP1 | ↑ | Mithocondrial function | HF, MI/R [143,144] |
| SENP2 | ↑ | Heart development and function | CHD [134] | |
| SENP5 | ↑ | Mithocondrial function | HF [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.-T.; Lee, A.; Kho, C. Ubiquitin and Ubiquitin-like Proteins in Cancer, Neurodegenerative Disorders, and Heart Diseases. Int. J. Mol. Sci. 2022, 23, 5053. https://doi.org/10.3390/ijms23095053
Hwang J-T, Lee A, Kho C. Ubiquitin and Ubiquitin-like Proteins in Cancer, Neurodegenerative Disorders, and Heart Diseases. International Journal of Molecular Sciences. 2022; 23(9):5053. https://doi.org/10.3390/ijms23095053
Chicago/Turabian StyleHwang, Jin-Taek, Ahyoung Lee, and Changwon Kho. 2022. "Ubiquitin and Ubiquitin-like Proteins in Cancer, Neurodegenerative Disorders, and Heart Diseases" International Journal of Molecular Sciences 23, no. 9: 5053. https://doi.org/10.3390/ijms23095053
APA StyleHwang, J.-T., Lee, A., & Kho, C. (2022). Ubiquitin and Ubiquitin-like Proteins in Cancer, Neurodegenerative Disorders, and Heart Diseases. International Journal of Molecular Sciences, 23(9), 5053. https://doi.org/10.3390/ijms23095053