
A2A Adenosine Receptor: A Possible Therapeutic Target for Alzheimer’s Disease by Regulating NLRP3 Inflammasome Activity?

 ,
,  ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Adenosine and A2A Receptor Activation
3. NLRP3 Inflammasome
4. Inflammasome and AD
5. Adenosine A2A Receptors and the Inflammasome in AD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schain, M.; Kreisl, W.C. Neuroinflammation in neurodegenerative disorders-a review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Cheng, X.; Zhong, S.; Liu, C.; Liu, F.; Zhao, C. Peripheral and central nervous system immune response crosstalk in amyotrophic lateral sclerosis. Front. Neurosci. 2020, 14, 575. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, F.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Simeone, P.; Lanuti, P.; Pieragostino, D.; Del Boccio, P.; Marchisio, M.; Miscia, S. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural. Regen. Res. 2020, 15, 850–856. [Google Scholar] [CrossRef]
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsy-chopharmacol. 2015, 25, 713–724. [Google Scholar] [CrossRef]
- Sami, N.; Rahman, S.; Kumar, V.; Zaidi, S.; Islam, A.; Ali, S.; Ahmad, F.; Hassan, M.I. Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 2017, 127, 1047–1057. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Blum, D.; Chern, Y.; Domenici, M.R.; Buée, L.; Lin, C.Y.; Rea, W.; Ferré, S.; Popoli, P. The role of adenosine tone and adenosine receptors in Huntington’s disease. J. Caffeine Adenosine Res. 2018, 8, 43–58. [Google Scholar] [CrossRef]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry. 2020, 25, 1876–1900. [Google Scholar] [CrossRef]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A adenosine receptors is reflected in platelets of patients with Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Aradi, S.D.; Hauser, R.A. Istradefylline—A first generation adenosine A(2A) antagonist for the treatment of Parkinson’s disease. Expert Rev. Neurother. 2021, 22, 317–333. [Google Scholar] [CrossRef]
- Du, H.; Tan, Y.; Li, C.H.; Zhao, Y.; Li, P.; Ning, Y.L.; Gao, R.B.; Wang, B.; Peng, Y.; Tan, S.W.; et al. High glutamate concentration reverses the inhibiitory effect of microglial adenosine 2° receptor on NLRP3 inflammasome assembly and activation. Neurosci. Lett. 2022, 769, 136431. [Google Scholar] [CrossRef]
- Zhao, W.; Ma, L.; Cai, C.; Gong, X. Caffeine Inhibits NLRP3 Inflammasome Activation by Suppressing MAPK/NF-kappaB and A2aR Signaling in LPS-Induced THP-1 Macrophages. Int. J. Biol. Sci. 2019, 15, 1571–1581. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Pérez-Torras, S. Who is who in adenosine transport. Front. Pharmacol. 2018, 9, 627. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a multi-signalling guardian angel in human diseases: When, Where and how does it exert its protective effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef] [Green Version]
- Kull, B.; Svenningsson, P.; Fredholm, B.B. Adenosine A2A receptors are colocalized with and activate g(olf) in rat striatum. Mol. Pharmacol. 2000, 58, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Schwindinger, W.F.; Mihalcik, L.J.; Giger, K.E.; Betz, K.S.; Stauffer, A.M.; Linden, J.; Herve, D.; Robishaw, J.D. Adenosine A2A receptor signaling and golf assembly show a specific requirement for the gamma7 subtype in the striatum. J. Biol. Chem. 2010, 285, 29787–29796. [Google Scholar] [CrossRef] [Green Version]
- Schulte, G.; Fredholm, B.B. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003, 15, 813–827. [Google Scholar] [CrossRef]
- Gessi, S.; Bencivenni, S.; Battistello, E.; Vincenzi, F.; Colotta, V.; Catarzi, D.; Varano, F.; Merighi, S.; Borea, P.A.; Varani, K. Inhibition of A2A Adenosine Receptor Signaling in Cancer Cells Proliferation by the Novel Antagonist TP455. Front. Pharmacol. 2017, 8, 888. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Gao, Z.G.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2020, 1–16. [Google Scholar] [CrossRef]
- Merighi, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Jacobson, K.A.; Gessi, S. A2A Adenosine Receptor Antagonists in Neurodegenerative Diseases. Curr. Med. Chem. 2021, in press. [Google Scholar] [CrossRef]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van der Jeugd, A. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry. 2016, 21, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Blum, D. Exacerbation of C1q dysregulation, synaptic loss and memory deficits in tau pathology linked to neuronal adenosine A2A receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive Oxygen Species at the Crossroads of Inflammasome and Inflammation. Front Physiol. 2014, 5, 352. [Google Scholar] [CrossRef]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Chong, L. NLRP3 Inflammasome and its Inhibitors: A Review. Front. Physiol. 2015, 5, 262. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Zhao, T.; Liu, M.; Cao, D.; Li, J.; Li, Y.; Xia, M.; Wang, X.; Zheng, T.; Liu, C.; et al. Targeting NLRP3 inflammasome in translational treatment of nervous system diseases: An update. Front. Pharmacol. 2021, 12, 707696. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, P.; Zhang, J.; Chen, W.; Gu, L. Silencing of the P2X7 receptor enhances amyloid-b phagocytosis by microglia biochemical and biophysical research. Communications. 2013, 434, 363–369. [Google Scholar] [CrossRef]
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation. 2006, 3, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, M.; O’Connor, J.J. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog. Brain Res. 2007, 163, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, O.J.; Srinivasan, N.; Pott, J.; Schiering, C.; Krausgruber, T.; Ilott, N.E.; Maloy, K.J. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3(+) Treg cell function in the intestine. Mucosal Immunol. 2015, 8, 1226–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holbrook, J.A.; Jarosz-Griffiths, H.H.; Caseley, E.; Lara-Reyna, S.; Poulter, J.A.; Williams-Gray, C.H.; Peckham, D.; McDermott, M.F. Neurodegenerative disease and the NLRP3 inflammasome. Front. Pharmacol. 2021, 12, 643254. [Google Scholar] [CrossRef]
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflammation. 2012, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nat. Cell Biol. 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.G.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as novel therapeutic strategies for NLRP3 inflammasome-related neurological, metabolic, and inflammatory diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Z.; Magupalli, V.G.; Pablo, J.L.; Dong, Y.; Vora, S.M.; Wang, L.; Fu, T.M.; Jacobson, M.P.; Greka, A.; et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 2021, 593, 607–611. [Google Scholar] [CrossRef]
- Rui, W.; Xiao, H.; Fan, Y.; Ma, Z.; Xiao, M.; Li, S.; Shi, J. Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer’s disease. J. Neuroinf. 2021, 18, 280. [Google Scholar] [CrossRef]
- Feng, Y.S.; Tan, Z.X.; Wu, L.Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of neurodegenerative diseases. Biomed. Pharmacother. 2021, 138, 111428. [Google Scholar] [CrossRef]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Attwell, D. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; Macleod, V.; Perrot, L.J.; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Blum-Degen, D.; Muller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Lynch, M.A. Neuroinflammatory changes negatively impact on LTP: A focus on IL-1beta. Brain Res. 2015, 1621, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Poloni, T.E.; Negro, G.; Varani, K.; Pasquini, S.; Vincenzi, F.; Borea, P.A.; Merighi, S. A2A adenosine receptor as a potential biomarker and a possible therapeutic target in Alzheimer’s disease. Cells 2021, 10, 2344. [Google Scholar] [CrossRef]
- Ising, C.; Heneka, M.T. Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration. Cell Death Dis. 2018, 9, 120. [Google Scholar] [CrossRef]
- Feng, Y.; Tan, Z.; Wu, L.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101192. [Google Scholar] [CrossRef]
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Golenbock, D.T. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar b-amyloid is attenuated by oligomeric b-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; Coleman, U.; Kingery-Gallagher, N.D.; El Khoury, J. Heterozygous CX3CR1 deficiency in microglia restores neuronal beta-amyloid clearance pathways and slows progression of Alzheimer’s like-disease in PS1-APP Mice. Front. Immunol. 2019, 10, 2780. [Google Scholar] [CrossRef] [Green Version]
- Beyer, M.M.S.; Lonnemann, N.; Remus, A.; Latz, E.; Heneka, M.T.; Korte, M. Enduring changes in neuronal function upon systemic inflammation are NLRP3 inflammasome dependent. J. Neurosci. 2020, 40, 5480–5494. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Heneka, M.T. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Daniels, M.J.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [Green Version]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Fiala, M.; Liu, Q.N.; Sayre, J.; Pop, V.; Brahmandam, V.; Graves, M.C.; Vinters, H.V. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur. J. Clin. Invest. 2002, 32, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezai-Zadeh, K.; Gate, D.; Town, T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J. Neuroimmune Pharmacol. 2009, 4, 462–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and Alzheimer’s disease. Neurobiol Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, T.C.; Hasegawa, Y.; Iguchi, R.; Cheung, A.; Caffrey, D.R.; Thatcher, E.J.; Golenbock, D.T. Inflammasome-derived cytokine IL18 suppresses amyloid-induced seizures in Alzheimer-prone mice. Proc. Natl. Acad. Sci. USA 2018, 115, 9002–9007. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.G.; Zhu, S.G.; Jones, R.A.; Griffin WS, T.; Mrak, R.E. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp. Neurol. 2000, 163, 388–391. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Karch, C.M.; Jeng, A.T.; Goate, A.M. Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J. Biol. Chem. 2012, 287, 42751–42762. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Holtzman, D.M. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Eacker, S.M.; Dawson, T.M.; Dawson, V.L. Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 2009, 10, 837–841. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by mi-croRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Wu, H.Z.; Ong, K.L.; Seeher, K.; Armstrong, N.J.; Thalamuthu, A.; Brodaty, H.; Schadev, P.; Mather, K. Circulating microRNAs as biomarkers of Alzheimer’s disease: A systematic review. J. Alzheimers Dis. 2015, 49, 755–766. [Google Scholar] [CrossRef]
- Lee, C.Y.; Ryu, I.S.; Ryu, J.H.; Cho, H.J. miRNAs as therapeutic tools in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 13012. [Google Scholar] [CrossRef]
- Watson, C.N.; Begum, G.; Ashman, E.; Thorn, D.; Yakoub, K.M.; Hariri, M.A.; Nehme, A.; Mondello, S.; Kobeissy, F.; Belli, A.; et al. Co-expression analysis of microRNAs and proteins in brain of Alzheimer’s disease patients. Cells 2022, 11, 163. [Google Scholar] [CrossRef]
- Mancuso, R.; Agostini, S.; Hernis, A.; Zanzottera, M.; Bianchi, A.; Clerici, M. Circulatory miR-223–3 p discriminates between Parkinson’s and Alzheimer’s Patients. Sci. Rep. 2019, 9, 9393. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, F.; Mancuso, R.; Agostini, S.; Piancone, F.; Marventano, I.; Saresella, M.; Hernis, A.; Fenoglio, C.; Galimberti, D.; Scarpini, E.; et al. Pharmacological and epigenetic regulators of NLRP3 inflammasome activation in Alzheimer’s disease. Pharmaceuticals 2021, 14, 1187. [Google Scholar] [CrossRef]
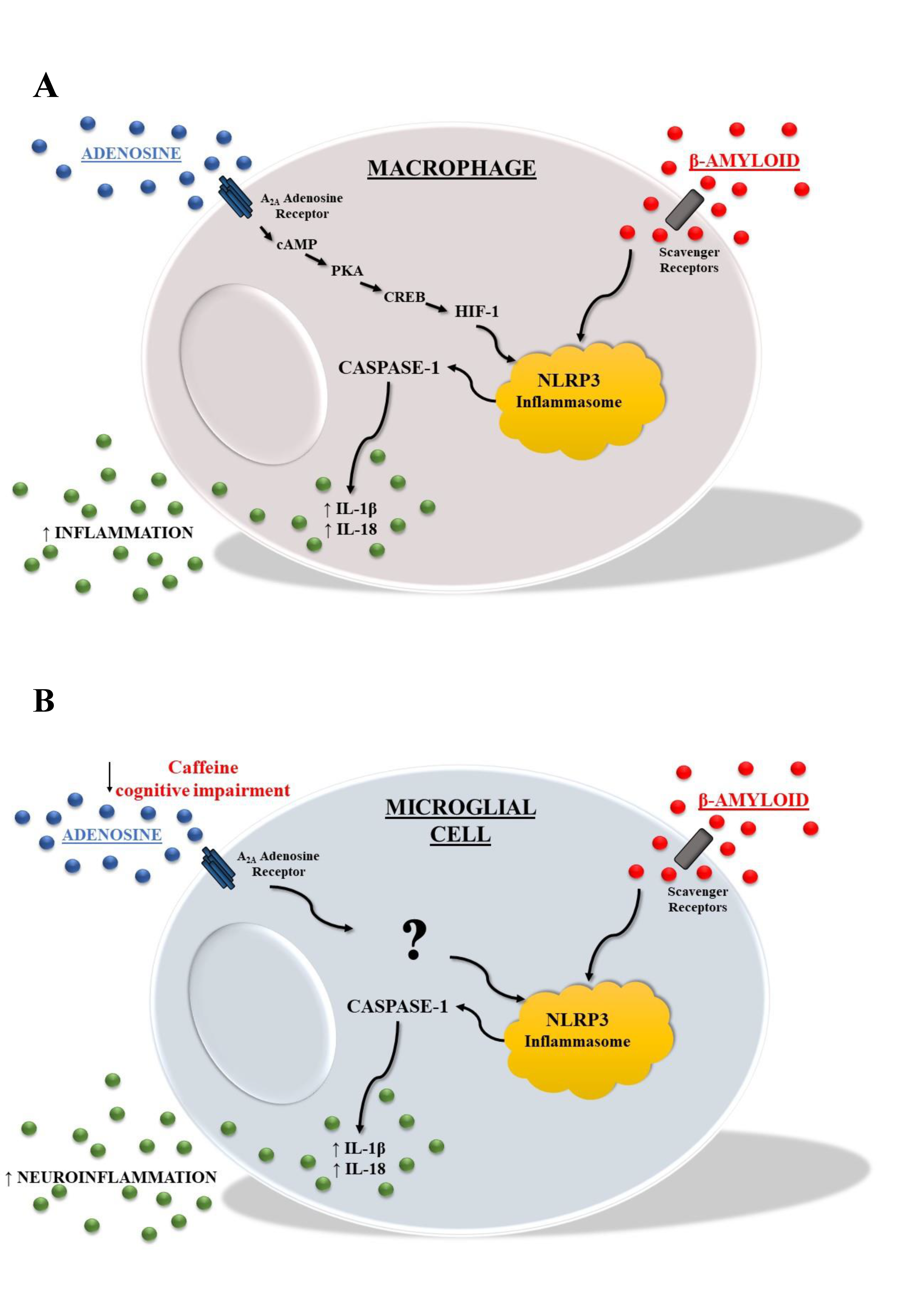
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine is required for sustained inflammasome activation via the A₂A receptor and the HIF-1α pathway. Nat. Commun. 2013, 4, 2909. [Google Scholar] [CrossRef] [Green Version]
- Kovács, E.G.; Alatshan, A.; Budai, M.M.; Czimmerer, Z.; Bíró, E.; Benkő, S. Caffeine has different immunomodulatory effect on the cytokine expression and NLRP3 inflammasome function in various human macrophage subpopulations. Nutrients 2021, 13, 2409. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, X.; Zhang, Y.; Liu, N.; Xue, X.; Fu, J. Caffeine treatment started before injury reduces hypoxic-ischemic white-matter damage in nenonatal rats by7 regulating phenotypic microglia polarization. Pediatr. Res. 2022, 1–12. [Google Scholar] [CrossRef]
- Deng, W. Neurobiology of injury to the developing brain. Nat. Rev. Neurol. 2010, 6, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.S.; Zhou, Y.G.; Li, W.; An, J.H.; Li, P.; Yang, N.; Chen, X.Y.; Xiong, R.P.; Liu, P.; Zaho, Y.; et al. Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. J. Neurosci. 2010, 30, 5802–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merighi, S.; Nigro, M.; Travagli, A.; Pasquini, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Gessi, S. A2A Adenosine Receptor: A Possible Therapeutic Target for Alzheimer’s Disease by Regulating NLRP3 Inflammasome Activity? Int. J. Mol. Sci. 2022, 23, 5056. https://doi.org/10.3390/ijms23095056
Merighi S, Nigro M, Travagli A, Pasquini S, Borea PA, Varani K, Vincenzi F, Gessi S. A2A Adenosine Receptor: A Possible Therapeutic Target for Alzheimer’s Disease by Regulating NLRP3 Inflammasome Activity? International Journal of Molecular Sciences. 2022; 23(9):5056. https://doi.org/10.3390/ijms23095056
Chicago/Turabian StyleMerighi, Stefania, Manuela Nigro, Alessia Travagli, Silvia Pasquini, Pier Andrea Borea, Katia Varani, Fabrizio Vincenzi, and Stefania Gessi. 2022. "A2A Adenosine Receptor: A Possible Therapeutic Target for Alzheimer’s Disease by Regulating NLRP3 Inflammasome Activity?" International Journal of Molecular Sciences 23, no. 9: 5056. https://doi.org/10.3390/ijms23095056
APA StyleMerighi, S., Nigro, M., Travagli, A., Pasquini, S., Borea, P. A., Varani, K., Vincenzi, F., & Gessi, S. (2022). A2A Adenosine Receptor: A Possible Therapeutic Target for Alzheimer’s Disease by Regulating NLRP3 Inflammasome Activity? International Journal of Molecular Sciences, 23(9), 5056. https://doi.org/10.3390/ijms23095056