
Evidence for Non-Essential Salt Bridges in the M-Gates of Mitochondrial Carrier Proteins

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
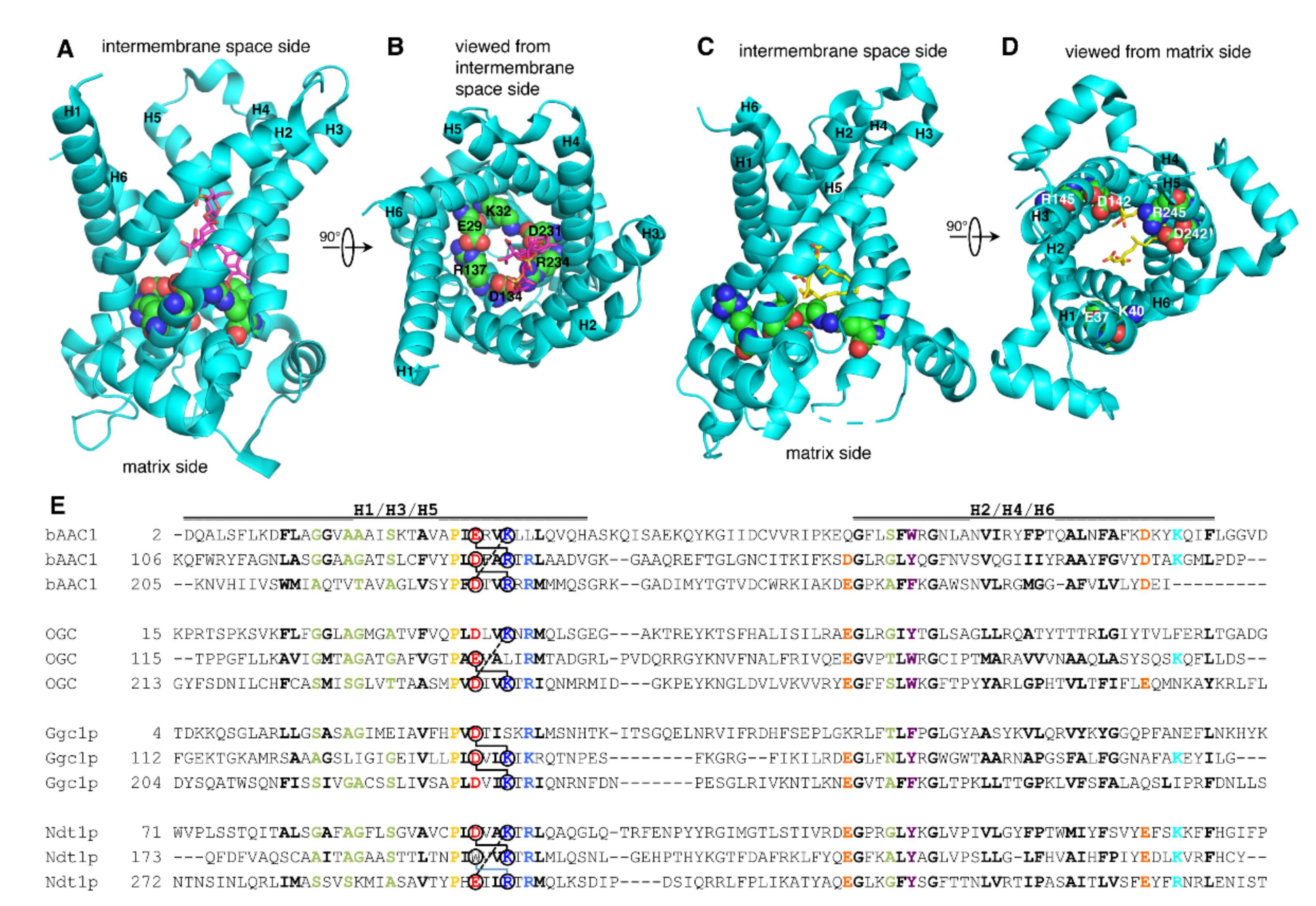
2.1. Structure and Sequence Analysis of the Matrix Salt Bridge Network of MCs
2.2. Effects on Activity by Mutating the Charged Residue Positions in the PX[DE]XX[KR] Motifs of OGC
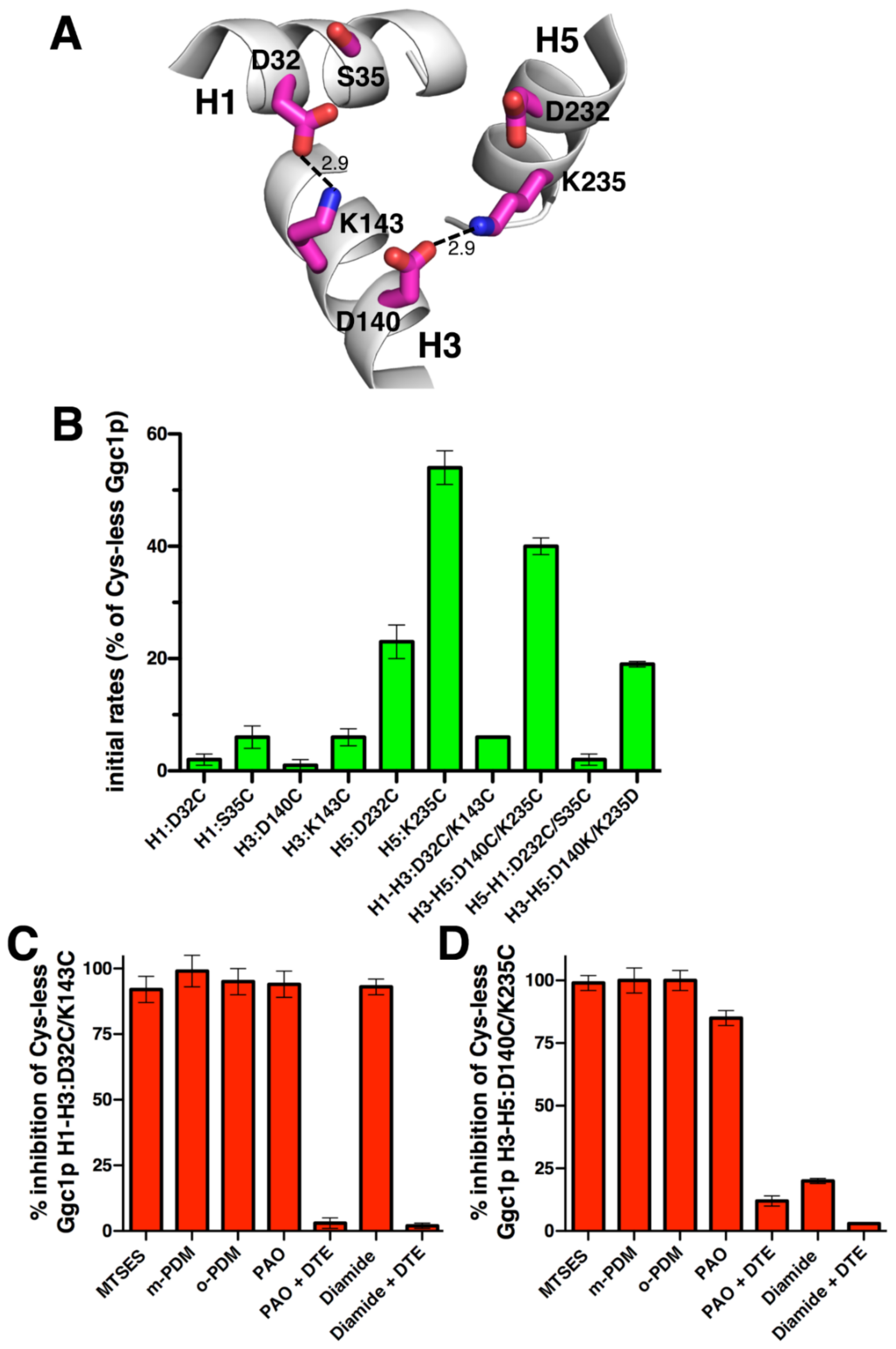
2.3. Effects on Activity by Mutating the Charged Residue Positions in the PX[DE]XX[KR] Motifs of Ggc1p
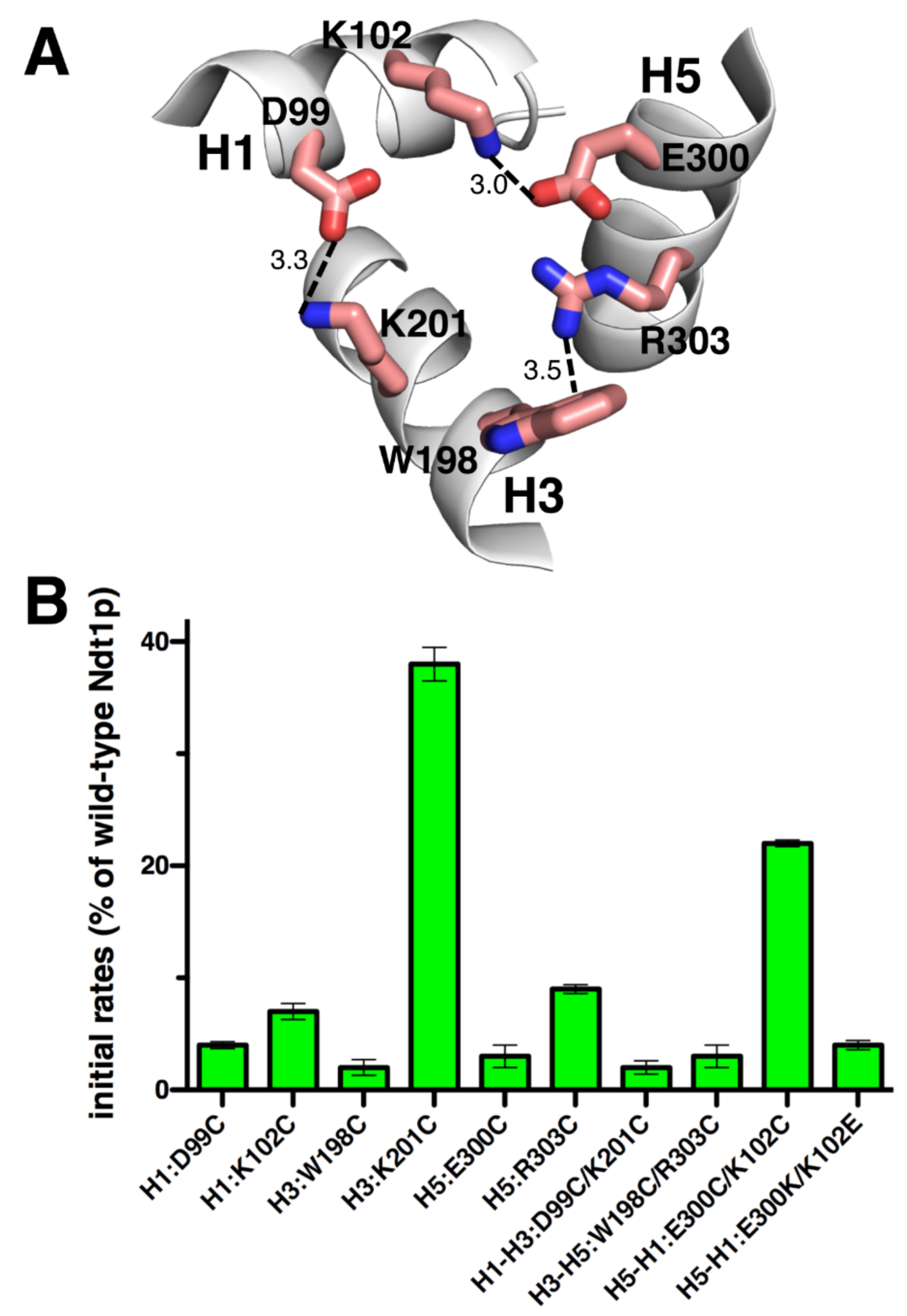
2.4. Effects on Activity by Mutating the Charged Residue Positions in the PX[DE]XX[KR] Motifs of Ndt1p
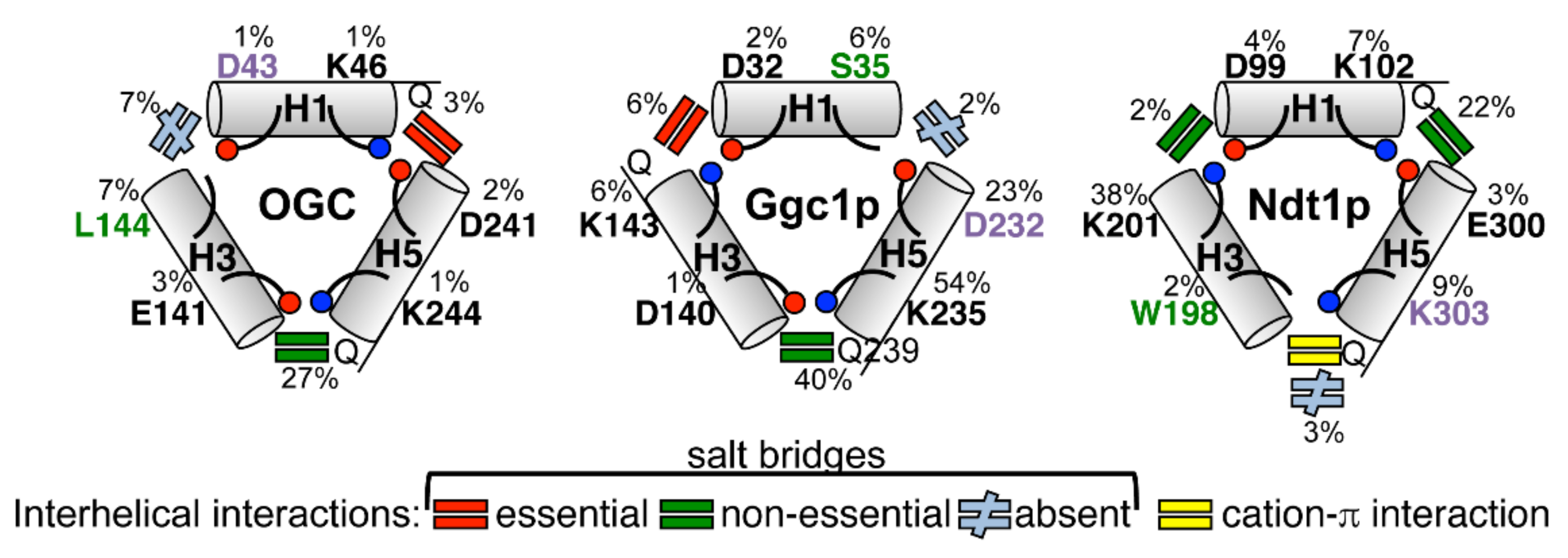
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Construction of Plasmids and Site-Directed Mutagenesis
4.3. Overexpression and Purification of the Recombinant MC Proteins
4.4. Reconstitution of the Purified MC Proteins into Liposomes and Transport Measurements
4.5. Additional Experimental Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Krämer, R.; Palmieri, F. Metabolite Carriers in Mitochondria. In Molecular Mechanisms in Bioenergetics; New Comprehensive Biochemistry; Ernster, L., Ed.; Elsevier: Amsterdam, The Netherlands, 1992; Volume 23, pp. 359–384. [Google Scholar]
- Palmieri, F. The Mitochondrial Transporter Family (SLC25): Physiological and Pathological Implications. Pflügers Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The Mitochondrial Transporter Family SLC25: Identification, Properties and Physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; Kunji, E.R.S. Structural Mechanism of Transport of Mitochondrial Carriers. Annu. Rev. Biochem. 2021, 90, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. Energy Metabolism | Mitochondrial Transporters of the Solute Carrier 25 (SLC25) Superfamily. In Encyclopedia of Biological Chemistry III, 3rd ed.; Jez, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 213–243. ISBN 978-0-12-822040-5. [Google Scholar]
- Palmieri, F.; Monné, M. Discoveries, Metabolic Roles and Diseases of Mitochondrial Carriers: A Review. Biochim. Biophys Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Monné, M.; Daddabbo, L.; Gagneul, D.; Obata, T.; Hielscher, B.; Palmieri, L.; Miniero, D.V.; Fernie, A.R.; Weber, A.P.M.; Palmieri, F. Uncoupling Proteins 1 and 2 (UCP1 and UCP2) from Arabidopsis Thaliana Are Mitochondrial Transporters of Aspartate, Glutamate, and Dicarboxylates. J. Biol. Chem. 2018, 293, 4213–4227. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, V.; Vozza, A.; Calcagnile, V.; Gorgoglione, R.; Arrigoni, R.; Fontanesi, F.; Marobbio, C.M.T.; Castegna, A.; Palmieri, F.; Palmieri, L. Molecular Identification and Functional Characterization of a Novel Glutamate Transporter in Yeast and Plant Mitochondria. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 1249–1258. [Google Scholar] [CrossRef]
- Gorgoglione, R.; Porcelli, V.; Santoro, A.; Daddabbo, L.; Vozza, A.; Monné, M.; Di Noia, M.A.; Palmieri, L.; Fiermonte, G.; Palmieri, F. The Human Uncoupling Proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) Transport Sulfur Oxyanions, Phosphate and Dicarboxylates. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 724–733. [Google Scholar] [CrossRef]
- Fiermonte, G.; Walker, J.E.; Palmieri, F. Abundant Bacterial Expression and Reconstitution of an Intrinsic Membrane-Transport Protein from Bovine Mitochondria. Biochem. J. 1993, 294, 293–299. [Google Scholar] [CrossRef]
- Palmieri, F.; Agrimi, G.; Blanco, E.; Castegna, A.; Di Noia, M.A.; Iacobazzi, V.; Lasorsa, F.M.; Marobbio, C.M.T.; Palmieri, L.; Scarcia, P.; et al. Identification of Mitochondrial Carriers in Saccharomyces Cerevisiae by Transport Assay of Reconstituted Recombinant Proteins. Biochim. Biophys. Acta 2006, 1757, 1249–1262. [Google Scholar] [CrossRef] [Green Version]
- Runswick, M.J.; Walker, J.E.; Bisaccia, F.; Iacobazzi, V.; Palmieri, F. Sequence of the Bovine 2-Oxoglutarate/Malate Carrier Protein: Structural Relationship to Other Mitochondrial Transport Proteins. Biochemistry 1990, 29, 11033–11040. [Google Scholar] [CrossRef]
- Vozza, A.; Blanco, E.; Palmieri, L.; Palmieri, F. Identification of the Mitochondrial GTP/GDP Transporter in Saccharomyces Cerevisiae. J. Biol. Chem. 2004, 279, 20850–20857. [Google Scholar] [CrossRef] [Green Version]
- Todisco, S.; Agrimi, G.; Castegna, A.; Palmieri, F. Identification of the Mitochondrial NAD+ Transporter in Saccharomyces Cerevisiae. J. Biol. Chem. 2006, 281, 1524–1531. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, M. The ADP and ATP Transport in Mitochondria and Its Carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. Diseases Caused by Defects of Mitochondrial Carriers: A Review. Biochim. Biophys. Acta 2008, 1777, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Palmieri, F. Antiporters of the Mitochondrial Carrier Family. Curr. Top. Membr. 2014, 73, 289–320. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial Transporters of the SLC25 Family and Associated Diseases: A Review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monné, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [Green Version]
- Saraste, M.; Walker, J.E. Internal Sequence Repeats and the Path of Polypeptide in Mitochondrial ADP/ATP Translocase. FEBS Lett. 1982, 144, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. Mitochondrial Carrier Proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Capobianco, L.; Brandolin, G.; Palmieri, F. Transmembrane Topography of the Mitochondrial Phosphate Carrier Explored by Peptide-Specific Antibodies and Enzymatic Digestion. Biochemistry 1991, 30, 4963–4969. [Google Scholar] [CrossRef]
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Dolce, V.; Fiermonte, G.; Iacobazzi, V.; Zara, V. Transmembrane Topology, Genes, and Biogenesis of the Mitochondrial Phosphate and Oxoglutarate Carriers. J. Bioenerg. Biomembr. 1993, 25, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, F.; Capobianco, L.; Brandolin, G.; Palmieri, F. Transmembrane Topography of the Mitochondrial Oxoglutarate Carrier Assessed by Peptide-Specific Antibodies and Enzymatic Cleavage. Biochemistry 1994, 33, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, L.; Bisaccia, F.; Michel, A.; Sluse, F.E.; Palmieri, F. The N- and C-Termini of the Tricarboxylate Carrier Are Exposed to the Cytoplasmic Side of the Inner Mitochondrial Membrane. FEBS Lett. 1995, 357, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of Mitochondrial ADP/ATP Carrier in Complex with Carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R.S. Structures of Yeast Mitochondrial ADP/ATP Carriers Support a Domain-Based Alternating-Access Transport Mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; King, M.S.; Zogg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.R.; Felix, C.M.; Swanson, J.M. Highly Conserved Charge-Pair Networks in the Mitochondrial Carrier Family. J. Mol. Biol. 1998, 277, 285–308. [Google Scholar] [CrossRef]
- Cappello, A.R.; Miniero, D.V.; Curcio, R.; Ludovico, A.; Daddabbo, L.; Stipani, I.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Functional and Structural Role of Amino Acid Residues in the Odd-Numbered Transmembrane Alpha-Helices of the Bovine Mitochondrial Oxoglutarate Carrier. J. Mol. Biol. 2007, 369, 400–412. [Google Scholar] [CrossRef]
- King, M.S.; Kerr, M.; Crichton, P.G.; Springett, R.; Kunji, E.R.S. Formation of a Cytoplasmic Salt Bridge Network in the Matrix State Is a Fundamental Step in the Transport Mechanism of the Mitochondrial ADP/ATP Carrier. Biochim. Biophys. Acta 2016, 1857, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial Carriers in the Cytoplasmic State Have a Common Substrate Binding Site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, M. The ADP, ATP Shuttle of the Mitochondrion. Trends Biochem. Sci. 1979, 4, 249–252. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. The Reconstituted Carnitine Carrier from Rat Liver Mitochondria: Evidence for a Transport Mechanism Different from That of the Other Mitochondrial Translocators. Biochim. Biophys. Acta 1994, 1189, 65–73. [Google Scholar] [CrossRef]
- Palmieri, F.; Pierri, C.L. Mitochondrial Metabolite Transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef]
- Kunji, E.R.S.; Aleksandrova, A.; King, M.S.; Majd, H.; Ashton, V.L.; Cerson, E.; Springett, R.; Kibalchenko, M.; Tavoulari, S.; Crichton, P.G.; et al. The Transport Mechanism of the Mitochondrial ADP/ATP Carrier. Biochim. Biophys. Acta 2016, 1863, 2379–2393. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Pierri, C.L. Structure and Function of Mitochondrial Carriers-Role of the Transmembrane Helix P and G Residues in the Gating and Transport Mechanism. FEBS Lett. 2010, 584, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Substrate Specificity of the Two Mitochondrial Ornithine Carriers Can Be Swapped by Single Mutation in Substrate Binding Site. J. Biol. Chem. 2012, 287, 7925–7934. [Google Scholar] [CrossRef] [Green Version]
- Stipani, V.; Cappello, A.R.; Daddabbo, L.; Natuzzi, D.; Miniero, D.V.; Stipani, I.; Palmieri, F. The Mitochondrial Oxoglutarate Carrier: Cysteine-Scanning Mutagenesis of Transmembrane Domain IV and Sensitivity of Cys Mutants to Sulfhydryl Reagents. Biochemistry 2001, 40, 15805–15810. [Google Scholar] [CrossRef]
- Cappello, A.R.; Curcio, R.; Miniero, D.V.; Stipani, I.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Functional and Structural Role of Amino Acid Residues in the Even-Numbered Transmembrane Alpha-Helices of the Bovine Mitochondrial Oxoglutarate Carrier. J. Mol. Biol. 2006, 363, 51–62. [Google Scholar] [CrossRef]
- Miniero, D.V.; Cappello, A.R.; Curcio, R.; Ludovico, A.; Daddabbo, L.; Stipani, I.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Functional and Structural Role of Amino Acid Residues in the Matrix α-Helices, Termini and Cytosolic Loops of the Bovine Mitochondrial Oxoglutarate Carrier. Biochim. Biophys. Acta 2011, 1807, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Dong, J.; Scott, R.A.; Ksenzenko, M.Y.; Rosen, B.P. The Role of Arsenic-Thiol Interactions in Metalloregulation of the Ars Operon. J. Biol. Chem. 1996, 271, 9291–9297. [Google Scholar] [CrossRef] [Green Version]
- Tonazzi, A.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by Site-Directed Mutagenesis and Chemical Modification of Three Vicinal Cysteine Residues in Rat Mitochondrial Carnitine/Acylcarnitine Transporter. J. Biol. Chem. 2005, 280, 19607–19612. [Google Scholar] [CrossRef] [Green Version]
- Akabas, M.H.; Stauffer, D.A.; Xu, M.; Karlin, A. Acetylcholine Receptor Channel Structure Probed in Cysteine-Substitution Mutants. Science 1992, 258, 307–310. [Google Scholar] [CrossRef]
- Briggs, C.; Mincone, L.; Wohlrab, H. Replacements of Basic and Hydroxyl Amino Acids Identify Structurally and Functionally Sensitive Regions of the Mitochondrial Phosphate Transport Protein. Biochemistry 1999, 38, 5096–5102. [Google Scholar] [CrossRef]
- Ma, C.; Remani, S.; Sun, J.; Kotaria, R.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. Identification of the Substrate Binding Sites within the Yeast Mitochondrial Citrate Transport Protein. J. Biol. Chem. 2007, 282, 17210–17220. [Google Scholar] [CrossRef] [Green Version]
- Müller, V.; Heidkämper, D.; Nelson, D.R.; Klingenberg, M. Mutagenesis of Some Positive and Negative Residues Occurring in Repeat Triad Residues in the ADP/ATP Carrier from Yeast. Biochemistry 1997, 36, 16008–16018. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C.; Palmieri, F. Site-Directed Mutagenesis of Charged Amino Acids of the Human Mitochondrial Carnitine/Acylcarnitine Carrier: Insight into the Molecular Mechanism of Transport. Biochim. Biophys. Acta 2010, 1797, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Tzagoloff, A.; Jang, J.; Glerum, D.M.; Wu, M. FLX1 Codes for a Carrier Protein Involved in Maintaining a Proper Balance of Flavin Nucleotides in Yeast Mitochondria. J. Biol. Chem. 1996, 271, 7392–7397. [Google Scholar] [CrossRef] [Green Version]
- Titus, S.A.; Moran, R.G. Retrovirally Mediated Complementation of the GlyB Phenotype. Cloning of a Human Gene Encoding the Carrier for Entry of Folates into Mitochondria. J. Biol. Chem. 2000, 275, 36811–36817. [Google Scholar] [CrossRef] [Green Version]
- Marobbio, C.M.T.; Di Noia, M.A.; Palmieri, F. Identification of a Mitochondrial Transporter for Pyrimidine Nucleotides in Saccharomyces Cerevisiae: Bacterial Expression, Reconstitution and Functional Characterization. Biochem. J. 2006, 393, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.; Favre, C.; Lasorsa, F.M.; Leahy, M.; Trigiante, G.; Stroebel, P.; Marx, A.; Loughran, G.; O’Callaghan, K.; Marobbio, C.M.T.; et al. The Insulin-like Growth Factor-I-MTOR Signaling Pathway Induces the Mitochondrial Pyrimidine Nucleotide Carrier to Promote Cell Growth. Mol. Biol. Cell 2007, 18, 3545–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, F.; Rieder, B.; Ventrella, A.; Blanco, E.; Do, P.T.; Nunes-Nesi, A.; Trauth, A.U.; Fiermonte, G.; Tjaden, J.; Agrimi, G.; et al. Molecular Identification and Functional Characterization of Arabidopsis Thaliana Mitochondrial and Chloroplastic NAD+ Carrier Proteins. J. Biol. Chem. 2009, 284, 31249–31259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiermonte, G.; Paradies, E.; Todisco, S.; Marobbio, C.M.T.; Palmieri, F. A Novel Member of Solute Carrier Family 25 (SLC25A42) Is a Transporter of Coenzyme A and Adenosine 3′,5′-Diphosphate in Human Mitochondria. J. Biol. Chem. 2009, 284, 18152–18159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrimi, G.; Russo, A.; Scarcia, P.; Palmieri, F. The Human Gene SLC25A17 Encodes a Peroxisomal Transporter of Coenzyme A, FAD and NAD+. Biochem. J. 2012, 443, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrimi, G.; Russo, A.; Pierri, C.L.; Palmieri, F. The Peroxisomal NAD+ Carrier of Arabidopsis Thaliana Transports Coenzyme A and Its Derivatives. J. Bioenerg. Biomem. 2012, 44, 333–340. [Google Scholar] [CrossRef]
- Bernhardt, K.; Wilkinson, S.; Weber, A.P.M.; Linka, N. A Peroxisomal Carrier Delivers NAD+ and Contributes to Optimal Fatty Acid Degradation during Storage Oil Mobilization. Plant J. 2012, 69, 1–13. [Google Scholar] [CrossRef]
- Di Noia, M.A.; Todisco, S.; Cirigliano, A.; Rinaldi, T.; Agrimi, G.; Iacobazzi, V.; Palmieri, F. The Human SLC25A33 and SLC25A36 Genes of Solute Carrier Family 25 Encode Two Mitochondrial Pyrimidine Nucleotide Transporters. J. Biol. Chem. 2014, 289, 33137–33148. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the Mitochondrial ATP-Mg/Pi Transporter. Bacterial Expression, Reconstitution, Functional Characterization, and Tissue Distribution. J. Biol. Chem. 2004, 279, 30722–30730. [Google Scholar] [CrossRef] [Green Version]
- Traba, J.; Satrústegui, J.; del Arco, A. Characterization of SCaMC-3-like/Slc25a41, a Novel Calcium-Independent Mitochondrial ATP-Mg/Pi Carrier. Biochem. J. 2009, 418, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Daddabbo, L.; Giannossa, L.C.; Nicolardi, M.C.; Palmieri, L.; Miniero, D.V.; Mangone, A.; Palmieri, F. Mitochondrial ATP-Mg/Phosphate Carriers Transport Divalent Inorganic Cations in Complex with ATP. J. Bioenerg. Biomembr. 2017, 49, 369–380. [Google Scholar] [CrossRef]
- Palmieri, L.; Palmieri, F.; Runswick, M.J.; Walker, J.E. Identification by Bacterial Expression and Functional Reconstitution of the Yeast Genomic Sequence Encoding the Mitochondrial Dicarboxylate Carrier Protein. FEBS Lett. 1996, 399, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Fiermonte, G.; Palmieri, L.; Dolce, V.; Lasorsa, F.M.; Palmieri, F.; Runswick, M.J.; Walker, J.E. The Sequence, Bacterial Expression, and Functional Reconstitution of the Rat Mitochondrial Dicarboxylate Transporter Cloned via Distant Homologs in Yeast and Caenorhabditis Elegans. J. Biol. Chem. 1998, 273, 24754–24759. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Picault, N.; Arrigoni, R.; Besin, E.; Palmieri, F.; Hodges, M. Molecular Identification of Three Arabidopsis Thaliana Mitochondrial Dicarboxylate Carrier Isoforms: Organ Distribution, Bacterial Expression, Reconstitution into Liposomes and Functional Characterization. Biochem. J. 2008, 410, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Vozza, A.; Agrimi, G.; De Marco, V.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the Yeast Mitochondrial Transporter for Oxaloacetate and Sulfate. J. Biol. Chem. 1999, 274, 22184–22190. [Google Scholar] [CrossRef] [Green Version]
- Marobbio, C.M.T.; Giannuzzi, G.; Paradies, E.; Pierri, C.L.; Palmieri, F. α-Isopropylmalate, a Leucine Biosynthesis Intermediate in Yeast, Is Transported by the Mitochondrial Oxalacetate Carrier. J. Biol. Chem. 2008, 283, 28445–28553. [Google Scholar] [CrossRef] [Green Version]
- Runswick, M.J.; Powell, S.J.; Nyren, P.; Walker, J.E. Sequence of the Bovine Mitochondrial Phosphate Carrier Protein: Structural Relationship to ADP/ATP Translocase and the Brown Fat Mitochondria Uncoupling Protein. EMBO J. 1987, 6, 1367–1373. [Google Scholar] [CrossRef]
- Dolce, V.; Iacobazzi, V.; Palmieri, F.; Walker, J.E. The Sequences of Human and Bovine Genes of the Phosphate Carrier from Mitochondria Contain Evidence of Alternatively Spliced Forms. J. Biol. Chem. 1994, 269, 10451–10460. [Google Scholar] [CrossRef]
- Wang, Y.; Tajkhorshid, E. Electrostatic Funneling of Substrate in Mitochondrial Inner Membrane Carriers. Proc. Natl. Acad. Sci. USA 2008, 105, 9598–9603. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, A.; Pierri, C.L.; Palmieri, F.; Klingenberg, M. The Switching Mechanism of the Mitochondrial ADP/ATP Carrier Explored by Free-Energy Landscapes. Biochim. Biophys. Acta 2016, 1857, 772–781. [Google Scholar] [CrossRef]
- Pasquadibisceglie, A.; Polticelli, F. Structural Determinants of Ligands Recognition by the Human Mitochondrial Basic Amino Acids Transporter SLC25A29. Insights from Molecular Dynamics Simulations of the c-State. Comput. Struct. Biotechnol. J. 2021, 19, 5600–5612. [Google Scholar] [CrossRef]
- Sahin-Tóth, M.; Dunten, R.L.; Gonzalez, A.; Kaback, H.R. Functional Interactions between Putative Intramembrane Charged Residues in the Lactose Permease of Escherichia Coli. Proc. Natl. Acad. Sci. USA 1992, 89, 10547–10551. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-Directed Mutagenesis by Overlap Extension Using the Polymerase Chain Reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Palmieri, F.; Indiveri, C.; Bisaccia, F.; Iacobazzi, V. Mitochondrial Metabolite Carrier Proteins: Purification, Reconstitution, and Transport Studies. Methods Enzymol. 1995, 260, 349–369. [Google Scholar] [CrossRef]
- Palmieri, L.; Lasorsa, F.M.; Iacobazzi, V.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the Mitochondrial Carnitine Carrier in Saccharomyces Cerevisiae. FEBS Lett. 1999, 462, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Vozza, A.; Hönlinger, A.; Dietmeier, K.; Palmisano, A.; Zara, V.; Palmieri, F. The Mitochondrial Dicarboxylate Carrier Is Essential for the Growth of Saccharomyces Cerevisiae on Ethanol or Acetate as the Sole Carbon Source. Mol. Microbiol. 1999, 31, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Castegna, A.; Scarcia, P.; Agrimi, G.; Palmieri, L.; Rottensteiner, H.; Spera, I.; Germinario, L.; Palmieri, F. Identification and Functional Characterization of a Novel Mitochondrial Carrier for Citrate and Oxoglutarate in S. Cerevisiae. J. Biol. Chem. 2010, 285, 17359–17370. [Google Scholar] [CrossRef] [Green Version]
- Poduri, A.; Heinzen, E.; Chitsazzadeh, V.; Lasorsa, F.; LaCoursiere, C.; Martin, E.; Yusakaitis, C.; Hill, R.; Elhosary, P.; Atabay, K.; et al. SLC25A22 Is a Novel Gene for Migrating Partial Seizures in Infancy. Ann. Neurol. 2013, 76, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Klingenberg, M. Direct Methods for Measuring Metabolite Transport and Distribution in Mitochondria. Methods Enzym. 1979, 56, 279–301. [Google Scholar]
- Fiermonte, G.; Dolce, V.; Palmieri, F. Expression in Escherichia Coli, Functional Characterization, and Tissue Distribution of Isoforms A and B of the Phosphate Carrier from Bovine Mitochondria. J. Biol. Chem. 1998, 273, 22782–22787. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Miniero, D.V.; Obata, T.; Daddabbo, L.; Palmieri, L.; Vozza, A.; Nicolardi, M.C.; Fernie, A.R.; Palmieri, F. Functional Characterization and Organ Distribution of Three Mitochondrial ATP-Mg/Pi Carriers in Arabidopsis Thaliana. Biochim. Biophys. Acta 2015, 1847, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, V.; Fiermonte, G.; Longo, A.; Palmieri, F. The Human Gene SLC25A29, of Solute Carrier Family 25, Encodes a Mitochondrial Transporter of Basic Amino Acids. J. Biol. Chem. 2014, 289, 13374–13384. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miniero, D.V.; Monné, M.; Di Noia, M.A.; Palmieri, L.; Palmieri, F. Evidence for Non-Essential Salt Bridges in the M-Gates of Mitochondrial Carrier Proteins. Int. J. Mol. Sci. 2022, 23, 5060. https://doi.org/10.3390/ijms23095060
Miniero DV, Monné M, Di Noia MA, Palmieri L, Palmieri F. Evidence for Non-Essential Salt Bridges in the M-Gates of Mitochondrial Carrier Proteins. International Journal of Molecular Sciences. 2022; 23(9):5060. https://doi.org/10.3390/ijms23095060
Chicago/Turabian StyleMiniero, Daniela Valeria, Magnus Monné, Maria Antonietta Di Noia, Luigi Palmieri, and Ferdinando Palmieri. 2022. "Evidence for Non-Essential Salt Bridges in the M-Gates of Mitochondrial Carrier Proteins" International Journal of Molecular Sciences 23, no. 9: 5060. https://doi.org/10.3390/ijms23095060