
MicroRNA-449a Inhibits Triple Negative Breast Cancer by Disturbing DNA Repair and Chromatid Separation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
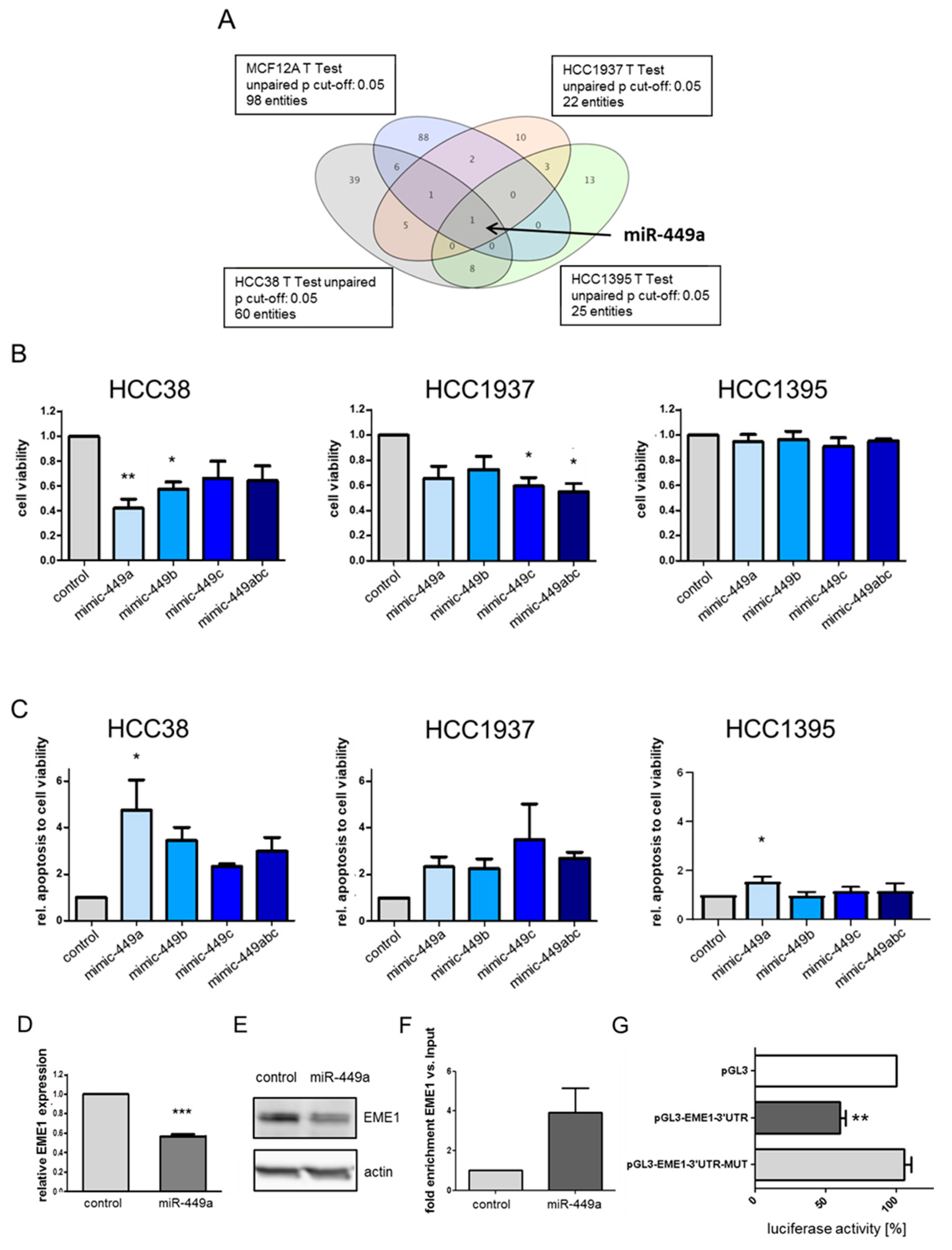
2.1. MiR-449a Is Epigenetically Silenced in TNBC and Re-Expression Leads to Reduced Cell Viability and Induced Apoptosis in TNBC Cell Lines
2.2. MiRNA-449a Deregulates Prominent Genes Involved in HDR and Directly Targets EME1
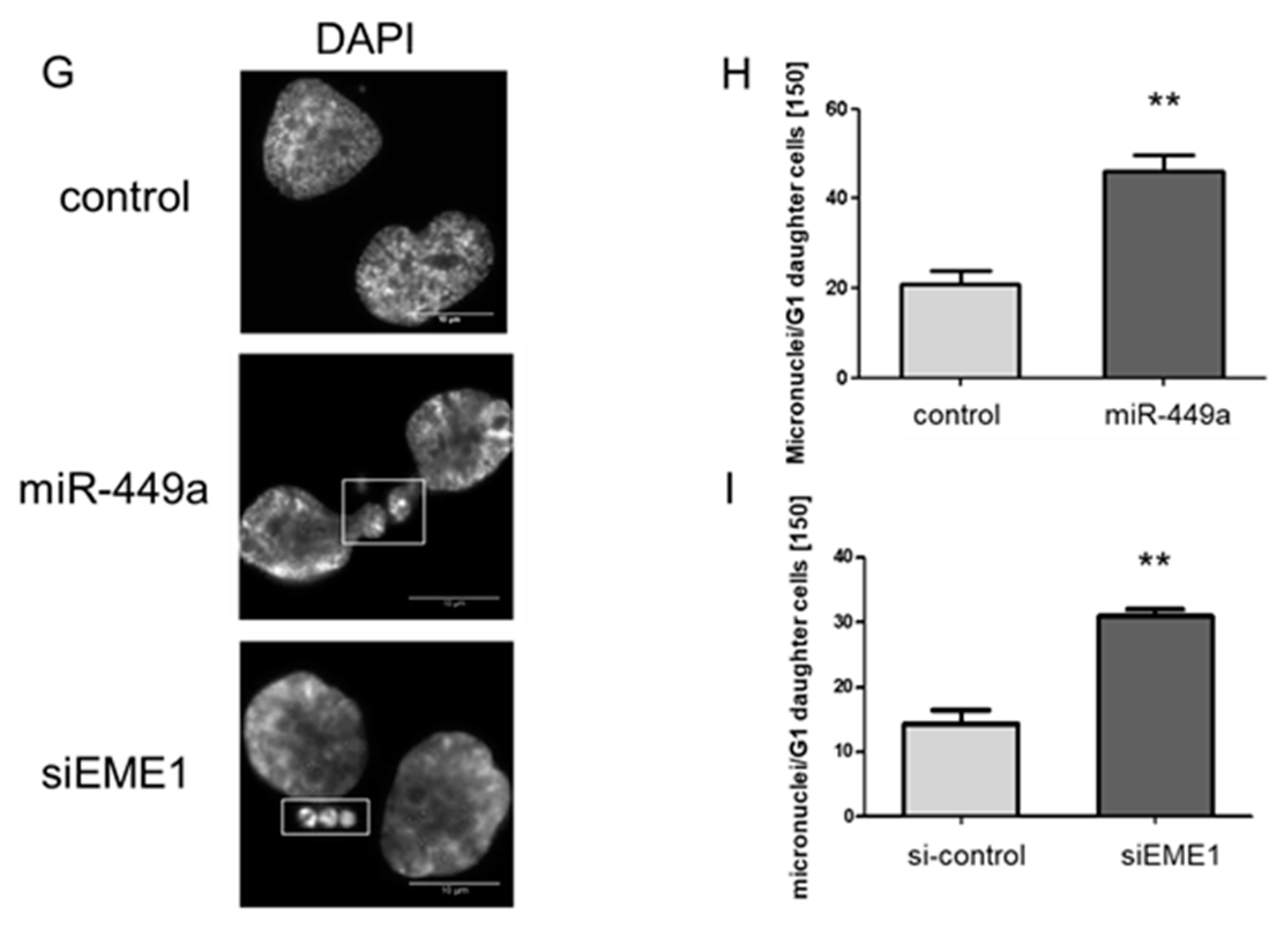
2.3. MiRNA-449a Leads to Incomplete Chromosome Segregation by Targeting EME1
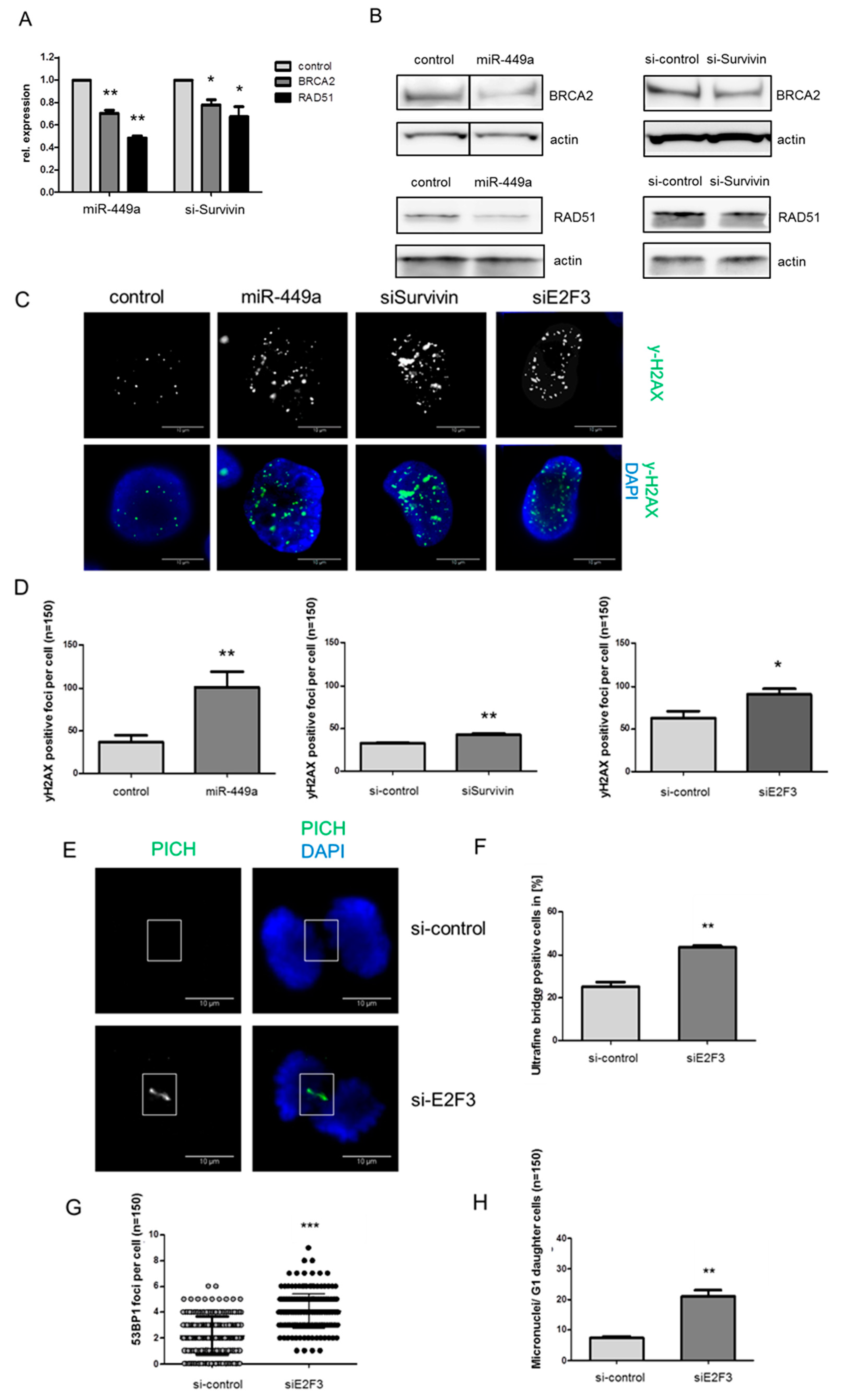
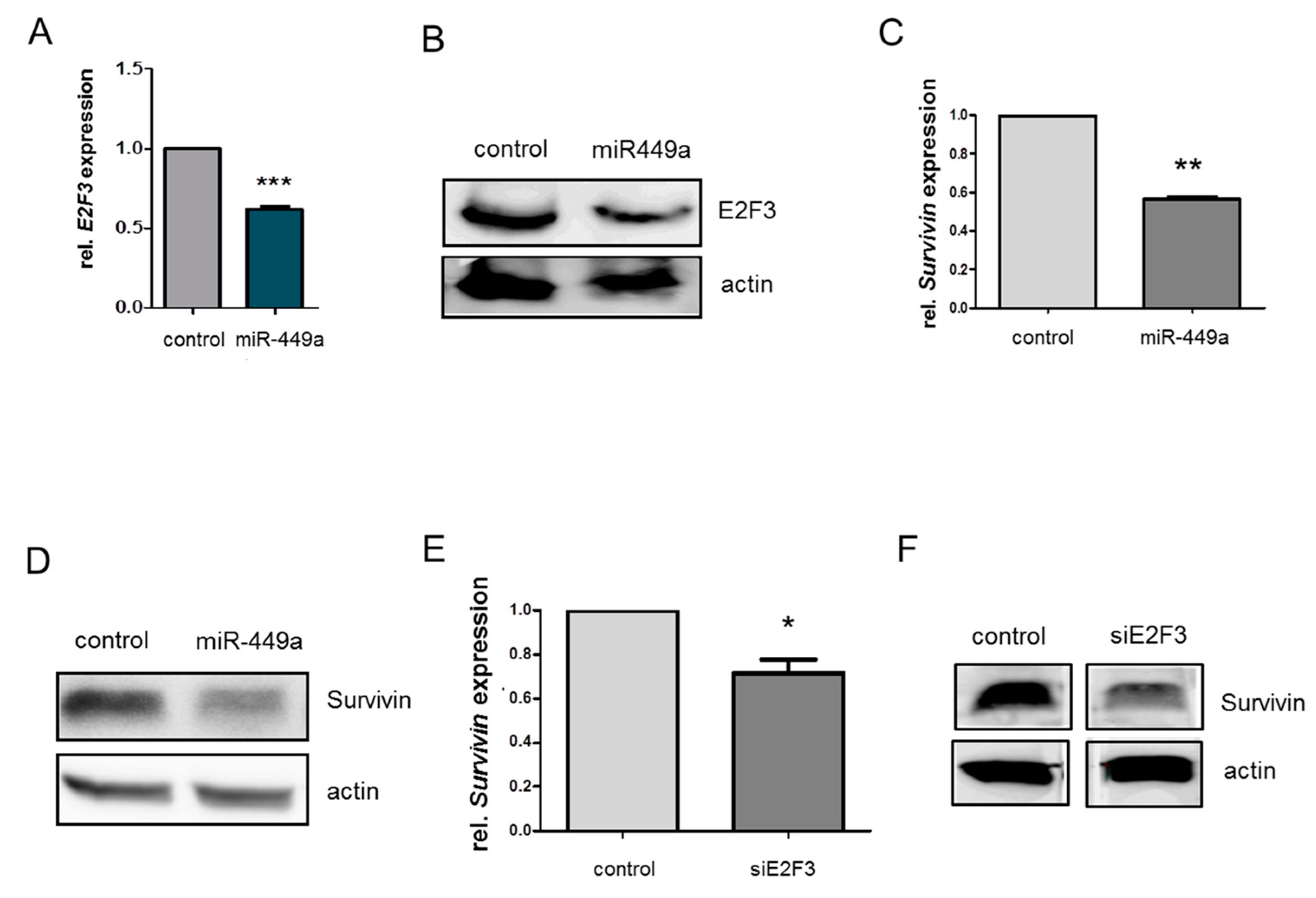
2.4. MiRNA-449a Reduces Homology-Directed Repair by Downregulation of E2F3 and Survivin
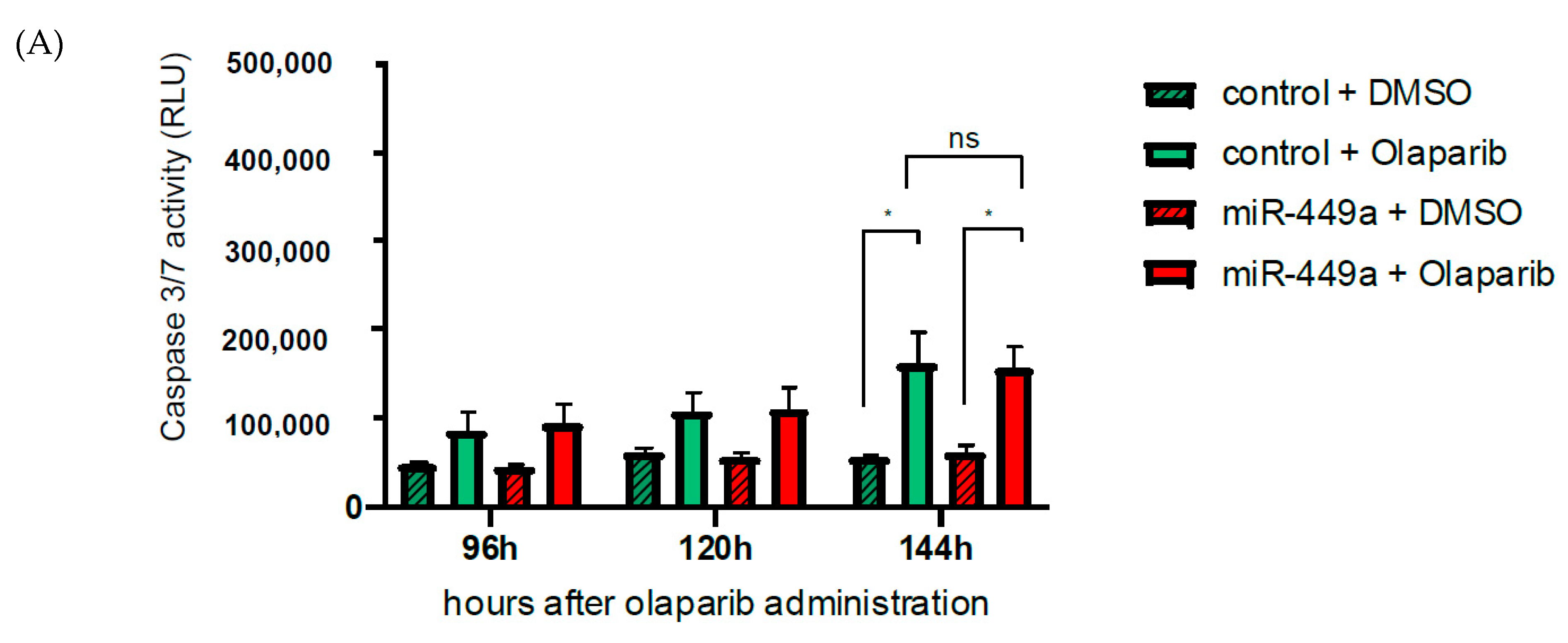
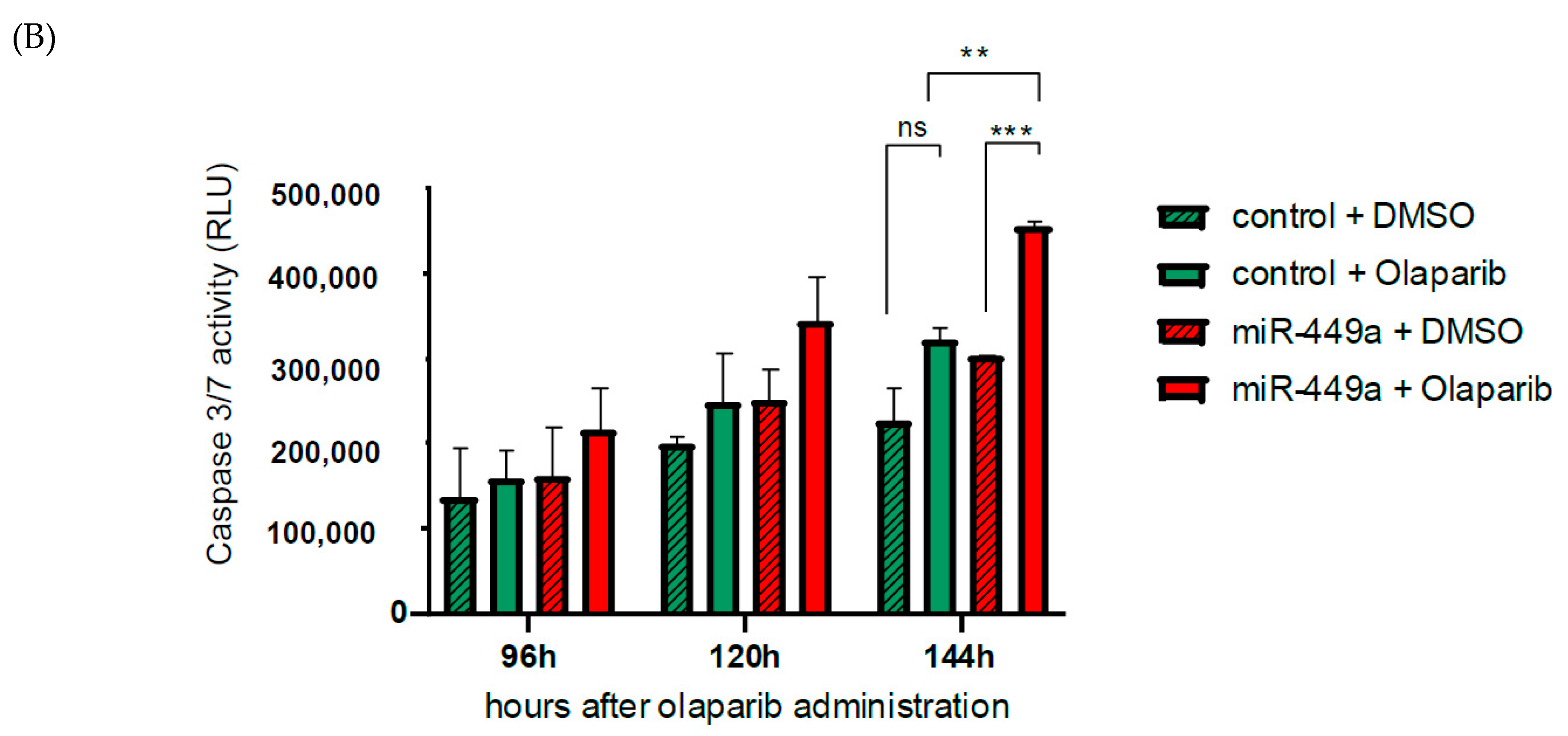
2.5. MiR-449a-Mediated Chromosomal Instability Increases the Toxic Effect of Olaparib in HCC1937 Cells
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stevens, K.N.; Vachon, C.M.; Couch, F.J. Genetic Susceptibility to Triple-Negative Breast Cancer. Cancer Res. 2013, 73, 2025–2030. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Birnbaum, M.J.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J. Virol. 1994, 68, 2521–2528. [Google Scholar] [CrossRef] [Green Version]
- Véquaud, E.; Desplanques, G.; Jézéquel, P.; Juin, P.; Barille-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2015, 155, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Weinandy, A.; Piroth, M.D.; Goswami, A.; Nolte, K.; Sellhaus, B.; Gerardo-Nava, J.; Eble, M.; Weinandy, S.; Cornelissen, C.; Clusmann, H.; et al. Cetuximab Induces Eme1-Mediated DNA Repair: A Novel Mechanism for Cetuximab Resistance. Neoplasia 2014, 16, 207–220.e4. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Naorem, L.D.; Muthaiyan, M.; Venkatesan, A. Identification of dysregulated miRNAs in triple negative breast cancer: A meta-analysis approach. J. Cell. Physiol. 2018, 234, 11768–11779. [Google Scholar] [CrossRef]
- Buurman, R.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Sandbothe, M.; Kreipe, H.; Wilkens, L.; Schlegelberger, B.; Kühnel, F.; Skawran, B. Histone Deacetylases Activate Hepatocyte Growth Factor Signaling by Repressing MicroRNA-449 in Hepatocellular Carcinoma Cells. Gastroenterology 2012, 143, 811–820.e15. [Google Scholar] [CrossRef]
- Sandbothe, M.; Buurman, R.; Reich, N.; Greiwe, L.; Vajen, B.; Gurlevik, E.; Schäffer, V.; Eilers, M.; Kühnel, F.; Vaquero, A.; et al. The microRNA-449 family inhibits TGF-beta-mediated liver cancer cell migration by targeting SOX4. J. Hepatol. 2017, 66, 1012–1021. [Google Scholar] [CrossRef]
- Ali, S.R.; Humphreys, K.J.; McKinnon, R.; Michael, M.Z. Impact of Histone Deacetylase Inhibitors on microRNA Expression and Cancer Therapy: A Review. Drug Dev. Res. 2015, 76, 296–317. [Google Scholar] [CrossRef]
- Vajen, B.; Greiwe, L.; Schäffer, V.; Eilers, M.; Huge, N.; Stalke, A.; Schlegelberger, B.; Illig, T.; Skawran, B. MicroRNA -192-5p inhibits migration of triple negative breast cancer cells and directly regulates Rho GTPase activating protein 19. Genes Chromosom. Cancer 2021, 60, 733–742. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Jézéquel, P.; Gouraud, W.; Ben Azzouz, F.; Guérin-Charbonnel, C.; Juin, P.P.; Lasla, H.; Campone, M. bc-GenExMiner 4.5: New mining module computes breast cancer differential gene expression analyses. Database 2021, 2021, 007. [Google Scholar] [CrossRef]
- National Cancer Institute. The Cancer Genome Atlas Program (TCGA). Available online: https://www.cancer.gov/tcga (accessed on 24 April 2020).
- Saal, L.H.; Vallon-Christersson, J.; Häkkinen, J.; Hegardt, C.; Grabau, D.; Winter, C.; Brueffer, C.; Tang, M.-H.E.; Reuterswärd, C.; Schulz, R.; et al. The Sweden Cancerome Analysis Network-Breast (SCAN-B) Initiative: A large-scale multicenter infrastructure towards implementation of breast cancer genomic analyses in the clinical routine. Genome Med. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Sørlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Gu, X.-D.; Li, Z.; Xiang, J.; Chen, Z. miR-449b inhibits the proliferation of SW1116 colon cancer stem cells through downregulation of CCND1 and E2F3 expression. Oncol. Rep. 2013, 30, 399–406. [Google Scholar] [CrossRef]
- Laulier, C.; Cheng, A.; Stark, J.M. The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res. 2011, 39, 5935–5944. [Google Scholar] [CrossRef] [Green Version]
- Sarbajna, S.; Davies, D.; West, S.C. Roles of SLX1–SLX4, MUS81–EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014, 28, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.-S.; Yin, M.-H.; Zhang, X.; Wang, Z.; Feng, S.-P.; Wang, G.-X.; Luo, Y.-J.; Liang, P.-Z.; Yang, X.-Q.; He, J.-X.; et al. Tumor-suppressive microRNA-449a induces growth arrest and senescence by targeting E2F3 in human lung cancer cells. Cancer Lett. 2014, 344, 195–203. [Google Scholar] [CrossRef]
- Geng, D.; Song, X.; Ning, F.; Song, Q.; Yin, H. MiR-34a Inhibits Viability and Invasion of Human Papillomavirus–Positive Cervical Cancer Cells by Targeting E2F3 and Regulating Survivin. Int. J. Gynecol. Cancer 2015, 25, 707–713. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortiner, P.; Swaisland, H.; Lau, A.; Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Marijon, H.; Lee, D.H.; Ding, L.-W.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vajen, B.; Bhowmick, R.; Greiwe, L.; Schäffer, V.; Eilers, M.; Reinkens, T.; Stalke, A.; Schmidt, G.; Fiedler, J.; Thum, T.; et al. MicroRNA-449a Inhibits Triple Negative Breast Cancer by Disturbing DNA Repair and Chromatid Separation. Int. J. Mol. Sci. 2022, 23, 5131. https://doi.org/10.3390/ijms23095131
Vajen B, Bhowmick R, Greiwe L, Schäffer V, Eilers M, Reinkens T, Stalke A, Schmidt G, Fiedler J, Thum T, et al. MicroRNA-449a Inhibits Triple Negative Breast Cancer by Disturbing DNA Repair and Chromatid Separation. International Journal of Molecular Sciences. 2022; 23(9):5131. https://doi.org/10.3390/ijms23095131
Chicago/Turabian StyleVajen, Beate, Rahul Bhowmick, Luisa Greiwe, Vera Schäffer, Marlies Eilers, Thea Reinkens, Amelie Stalke, Gunnar Schmidt, Jan Fiedler, Thomas Thum, and et al. 2022. "MicroRNA-449a Inhibits Triple Negative Breast Cancer by Disturbing DNA Repair and Chromatid Separation" International Journal of Molecular Sciences 23, no. 9: 5131. https://doi.org/10.3390/ijms23095131
APA StyleVajen, B., Bhowmick, R., Greiwe, L., Schäffer, V., Eilers, M., Reinkens, T., Stalke, A., Schmidt, G., Fiedler, J., Thum, T., DeLuca, D. S., Hickson, I. D., Schlegelberger, B., Illig, T., & Skawran, B. (2022). MicroRNA-449a Inhibits Triple Negative Breast Cancer by Disturbing DNA Repair and Chromatid Separation. International Journal of Molecular Sciences, 23(9), 5131. https://doi.org/10.3390/ijms23095131