
Novel Models of Crohn’s Disease Pathogenesis Associated with the Occurrence of Mitochondrial Dysfunction in Intestinal Cells

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Etiology of Crohn’s Disease
2.1. Diet
2.2. Smoking
2.3. The Effect of Therapeutics on CD Development
2.4. Genetic Factors
2.5. Dysbiosis
3. Models of CD Pathogenesis Based on Mitochondrial Dysfunction
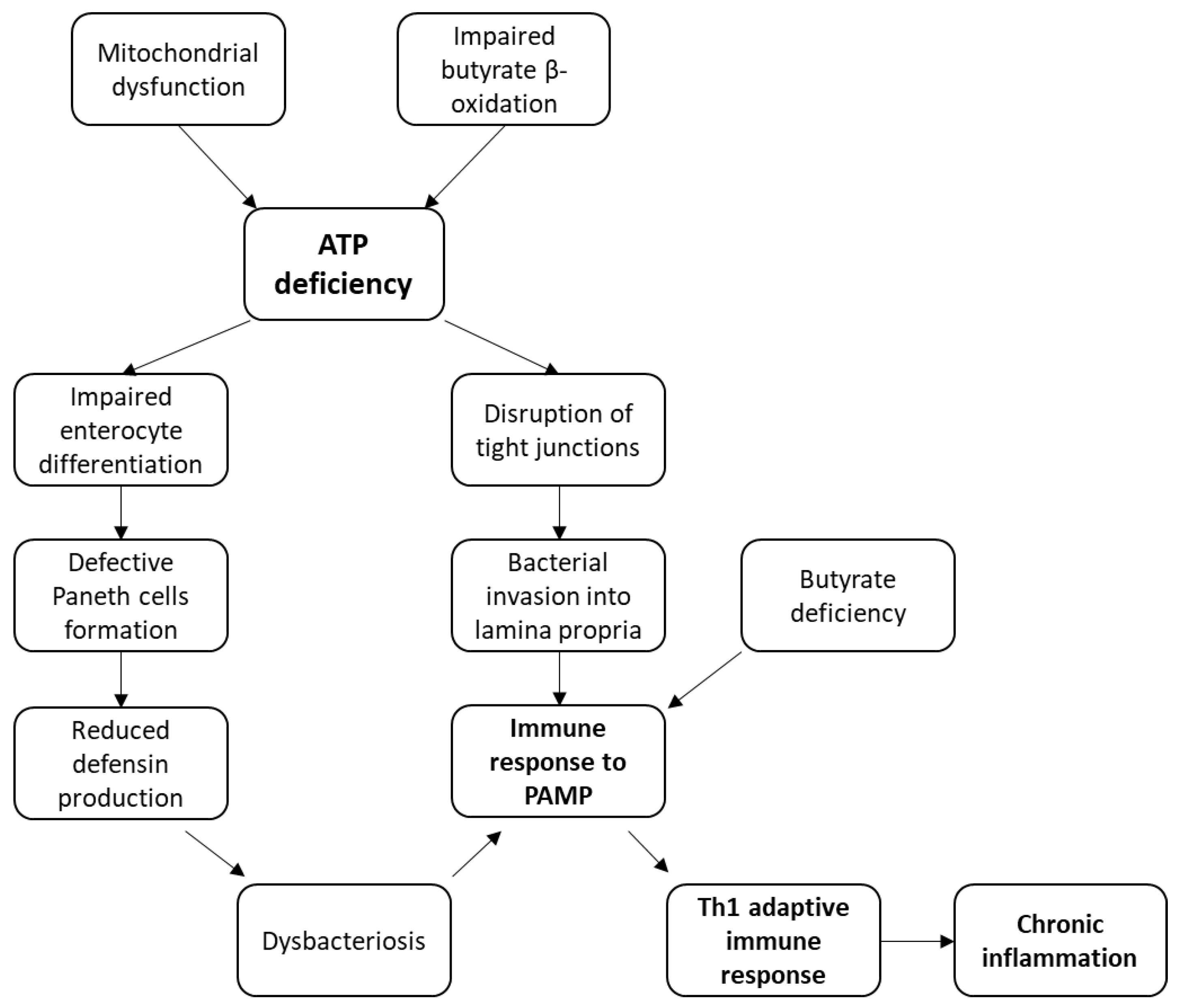
3.1. Models of CD Pathogenesis Based on an Energy Deficit in the Intestine Cells
3.2. Mechanism of CD Pathogenesis Based on Impaired Enterocyte Differentiation
3.3. Mechanism of CD Pathogenesis Based on the Disruption of Tight Junction Integrity
3.4. Mechanism of CD Pathogenesis Based on Impaired Butyrate Oxidation and Its Deficiency
3.5. Immune Response to Bacterial Antigens in CD
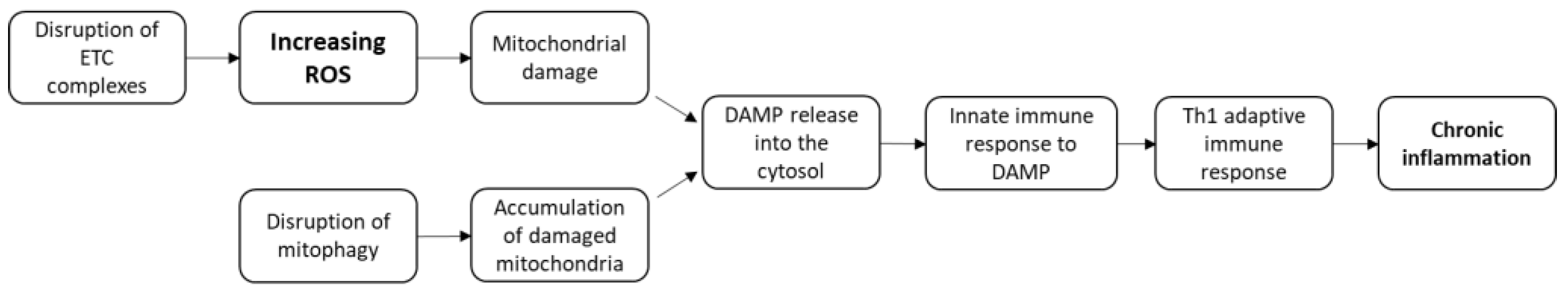
3.6. Models of CD Pathogenesis Based on ROS Production and Mitophagy Disorders
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet. Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Crohn’s Disease Complications. Date Views. Available online: www.webmd.com/ibd-crohns-disease/crohns-disease/crohns-disease-complications (accessed on 27 April 2021).
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic. Dis. 2020, 11, 2040622319899297. [Google Scholar] [CrossRef] [PubMed]
- Mikocka-Walus, A.; Pittet, V.; Rossel, J.-B.; von Känel, R.; Anderegg, C.; Bauerfeind, P.; Beglinger, C.; Begré, S.; Belli, D.; Bengoa, J.M.; et al. Symptoms of Depression and Anxiety Are Independently Associated with Clinical Recurrence of Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 829–835.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, W.; Goethel, A.; Bedrani, L.; Croitoru, M.K. Determinants of IBD Heritability: Genes, Bugs, and More. Inflamm. Bowel Dis. 2018, 24, 1133–1148. [Google Scholar] [CrossRef] [Green Version]
- Sartor, R.B. Mechanisms of Disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- Sidarala, V.; Pearson, G.L.; Parekh, V.S.; Thompson, B.; Christen, L.; Gingerich, M.A.; Zhu, J.; Stromer, T.; Ren, J.; Reck, E.C.; et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020, 5, e141138. [Google Scholar] [CrossRef]
- Wang, G.; Yang, Y.; Ma, H.; Shi, L.; Jia, W.; Hao, X.; Liu, W. LncRNA FENDRR Inhibits ox-LDL Induced Mitochondrial Energy Metabolism Disorder in Aortic Endothelial Cells via miR-18a-5p/PGC-1α Signaling Pathway. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Ray, K. Mitochondrial dysfunction in Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 260. [Google Scholar] [CrossRef]
- Carrière, J.; Darfeuille-Michaud, A.; Nguyen, H.T.T. Infectious etiopathogenesis of Crohn’s disease. World J. Gastroenterol. 2014, 20, 12102–12117. [Google Scholar] [CrossRef]
- Chapman-Kiddell, C.A.; Davies, P.S.; Gillen, L.; Radford-Smith, G.L. Role of diet in the development of inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 137–151. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Bugging of the Intestinal Mucosa. N. Engl. J. Med. 2007, 357, 708–710. [Google Scholar] [CrossRef] [Green Version]
- Geerling, B.J.; Badart-Smook, A.; Stockbrügger, R.W.; Brummer, R.J. Comprehensive nutritional status in recently diagnosed patients with inflammatory bowel disease compared with population controls. Eur. J. Clin. Nutr. 2000, 54, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Shoda, R.; Matsueda, K.; Yamato, S.; Umeda, N. Epidemiologic analysis of Crohn disease in Japan: Increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am. J. Clin. Nutr. 1996, 63, 741–745. [Google Scholar] [CrossRef]
- Seksik, P.; Nion-Larmurier, I.; Sokol, H.; Beaugerie, L.; Cosnes, J. Effects of light smoking consumption on the clinical course of Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 734–741. [Google Scholar] [CrossRef]
- Sutherland, L.R.; Ramcharan, S.; Bryant, H.; Fick, G. Effect of Cigarette Smoking on Recurrence of Crohn’s Disease. Gastroenterology 1990, 98, 1123–1128. [Google Scholar] [CrossRef]
- Parsi, M.A.; Achkar, J.; Richardson, S.; Katz, J.; Hammel, J.P.; Lashner, B.A.; Brzezinski, A. Predictors of response to infliximab in patients with Crohn’s disease. Gastroenterology 2002, 123, 707–713. [Google Scholar] [CrossRef]
- Cosnes, J.; Beaugerie, L.; Carbonnel, F.; Gendre, J. Smoking cessation and the course of Crohn’s disease: An intervention study. Gastroenterology 2001, 120, 1093–1099. [Google Scholar] [CrossRef]
- Parkes, G.C.; Whelan, K.; Lindsay, J.O. Smoking in inflammatory bowel disease: Impact on disease course and insights into the aetiology of its effect. J. Crohn’s Colitis 2014, 8, 717–725. [Google Scholar] [CrossRef]
- Benjamin, J.L.; Hedin, C.R.; Koutsoumpas, A.; Ng, S.C.; McCarthy, N.E.; Prescott, N.J.; Pessoa-Lopes, P.; Mathew, C.G.; Sanderson, J.; Hart, A.L.; et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm. Bowel Dis. 2012, 18, 1092–1100. [Google Scholar] [CrossRef]
- Benoni, C.; Prytz, H. Effects of smoking on the urine excretion of oral51Cr EDTA in ulcerative colitis. Gut 1998, 42, 656–658. [Google Scholar] [CrossRef] [Green Version]
- Verschuere, S.; Bracke, K.; Demoor, T.; Plantinga, M.; Verbrugghe, P.; Ferdinande, L.; Lambrecht, B.N.; Brusselle, G.; Cuvelier, C.A. Cigarette smoking alters epithelial apoptosis and immune composition in murine GALT. Lab. Investig. 2011, 91, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Frolkis, A.; A Dieleman, L.; Barkema, H.W.; Panaccione, R.; Ghosh, S.; Fedorak, R.N.; Madsen, K.; Kaplan, G.G.; on behalf of the Alberta IBD Consortium. Environment and the Inflammatory Bowel Diseases. Can. J. Gastroenterol. 2013, 27, e18–e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, H.; Malmborg, P.; Askling, J.; Ekbom, A.; Montgomery, S.M. Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scand. J. Gastroenterol. 2008, 43, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Cornish, J.; Tan, E.; Simillis, C.; Clark, S.K.; Teare, J.; Tekkis, P.P. The Risk of Oral Contraceptives in the Etiology of Inflammatory Bowel Disease: A Meta-Analysis. Am. J. Gastroenterol. 2008, 103, 2394–2400. [Google Scholar] [CrossRef]
- Cutolo, M.; Capellino, S.; Sulli, A.; Serioli, B.; Secchi, M.E.; Villaggio, B.; Straub, R.H. Estrogens and Autoimmune Diseases. Ann. N. Y. Acad. Sci. 2006, 1089, 538–547. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Higuchi, L.M.; Huang, E.S.; Khalili, H.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Aspirin, nonsteroidal an-ti-inflammatory drug use, and risk for Crohn disease and ulcerative colitis: A cohort study. Ann. Intern. Med. 2012, 156, 350–359. [Google Scholar] [CrossRef]
- Takeuchi, K.; Smale, S.; Premchand, P.; Maiden, L.; Sherwood, R.; Thjodleifsson, B.; Bjornsson, E.; Bjarnason, I. Prevalence and Mechanism of Nonsteroidal Anti-Inflammatory Drug–Induced Clinical Relapse in Patients With Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2006, 4, 196–202. [Google Scholar] [CrossRef]
- Brant, S.R. Update on the heritability of inflammatory bowel disease: The importance of twin studies. Inflamm. Bowel Dis. 2011, 17, 1–5. [Google Scholar] [CrossRef]
- Eckmann, L.; Karin, M. NOD2 and Crohn’s disease: Loss or gain of function? Immunity 2005, 22, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Lavrinenko, V.A.; Takun, K.V.; Minakovskaya, N.V.; Prudnikov, D.V.; Bydanov, O.I.; Aleinikova, O.V. Polymorphism of the NOD2/CARD15 gene is associated with an increased risk of developing a common chronic and steroid-resistant graft-versus-host reaction after allogeneic THSC. Transfusiology East. Eur. 2016, 3, 350–360. [Google Scholar]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2006, 39, 207–211. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Goll, M.G.; Halpern, M.E. DNA Methylation in Zebrafish. Prog. Mol. Biol Transl. Sci. 2011, 101, 193–218. [Google Scholar] [CrossRef] [Green Version]
- Dave, M.; Higgins, P.D.; Middha, S.; Rioux, K.P. The human gut microbiome: Current knowledge, challenges, and future directions. Transl. Res. 2012, 160, 246–257. [Google Scholar] [CrossRef]
- Hoyles, L.; Swann, J. Influence of the Human Gut Microbiome on the Metabolic Phenotype. In The Handbook of Metabolic Phenotyping, 1st ed.; Lindon, J., Nicholson, J., Holmes, E., Eds.; Elsevier: London, UK, 2019; pp. 535–560. [Google Scholar] [CrossRef]
- Aldars-García, L.; Marin, A.C.; Chaparro, M.; Gisbert, J.P. The Interplay between Immune System and Microbiota in Inflammatory Bowel Disease: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 3076. [Google Scholar] [CrossRef]
- O'Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Ullah, N.; Zha, L.; Bai, Y.; Khan, A.; Zhao, T.; Che, T.; Zhang, C. Alteration of Gut Microbiota in Inflammatory Bowel Disease (IBD): Cause or Consequence? IBD Treatment Targeting the Gut Microbiome. Pathogens 2019, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Sanchis-Artero, L.; Martínez-Blanch, J.F.; Manresa-Vera, S.; Cortés-Castell, E.; Valls-Gandia, M.; Iborra, M.; Paredes-Arquiola, J.M.; Boscá-Watts, M.; Huguet, J.M.; Gil-Borrás, R.; et al. Evaluation of changes in intestinal microbiota in Crohn’s disease patients after anti-TNF alpha treatment. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Chervy, M.; Barnich, N.; Denizot, J. Adherent-Invasive E. coli: Update on the Lifestyle of a Troublemaker in Crohn’s Disease. Int. J. Mol. Sci. 2020, 21, 3734. [Google Scholar] [CrossRef]
- Nicolson, G.L. Mitochondrial dysfunction and chronic disease: Treatment with natural supplements. Integr. Med. 2014, 13, 35–43. [Google Scholar]
- Sobenin, I.; Sazonova, M.; Postnov, A.; Bobryshev, Y.V.; Orekhov, A. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Restivo, N.L.; Srivastava, M.D.; Schafer, I.A.; Hoppel, C.L. Mitochondrial Dysfunction in a Patient with Crohn Disease: Possible Role in Pathogenesis. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef] [Green Version]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cell α-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [Green Version]
- Khaloian, S.; Rath, E.; Hammoudi, N.; Gleisinger, E.; Blutke, A.; Giesbertz, P.; Berger, E.; Metwaly, A.; Waldschmitt, N.; Allez, M.; et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 2020, 69, 1939–1951. [Google Scholar] [CrossRef]
- Bär, F.; Bochmann, W.; Widok, A.; von Medem, K.; Pagel, R.; Hirose, M.; Yu, X.; Kalies, K.; König, P.; Böhm, R.; et al. Mitochondrial Gene Polymorphisms That Protect Mice from Colitis. Gastroenterology 2013, 145, 1055–1063.e3. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Hagen, S.; Pothoulakis, C.; Chen, M.; Medina, N.; Warny, M.; LaMont, J. Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology 2000, 119, 139–150. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Santhanam, S.; Venkatraman, A.; Ramakrishna, B.S. Impairment of mitochondrial acetoacetyl CoA thiolase activity in the colonic mucosa of patients with ulcerative colitis. Gut 2007, 56, 1543–1549. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W.E.; Nance, S. Metabolic induction of experimental ulcerative colitis by inhibition of fatty acid oxidation. Br. J. Exp. Pathol. 1986, 67, 773–782. [Google Scholar]
- Shekhawat, P.S.; Srinivas, S.R.; Matern, D.; Bennett, M.J.; Boriack, R.; George, V.; Xu, H.; Prasad, P.D.; Roon, P.; Ganapathy, V. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (OCTN2(-/-)) mice. Mol. Genet. Metab. 2007, 92, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [Green Version]
- Cobo, E.; Kissoon-Singh, V.; Moreau, F.; Holani, R.; Chadee, K. MUC2 Mucin and Butyrate Contribute to the Synthesis of the Antimicrobial Peptide Cathelicidin in Response to Entamoeba histolytica- and Dextran Sodium Sulfate-Induced Colitis. Infect. Immun. 2017, 85, e00905-16. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, H.N.; Moroney, J.B.; Gan, H.; Shen, T.; Im, J.L.; Li, T.; Taylor, J.R.; Zan, H.; Casali, P. B cell-intrinsic epigenetic modulation of antibody responses by dietary fiber-derived short-chain fatty acids. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Hakansson, A.; Molin, G. Gut microbiota and inflammation. Nutrients 2011, 3, 637–682. [Google Scholar] [CrossRef]
- Newberry, R.; Lorenz, R.G. Organizing a mucosal defense. Immunol. Rev. 2005, 206, 6–21. [Google Scholar] [CrossRef]
- Yamamoto-Furusho, J.K.; Sanchez-Muñoz, F.; Dominguez-Lopez, A. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Dang, P.M.; Dewas, C.; Gaudry, M.; Fay, M.; Pedruzzi, E.; Gougerot-Pocidalo, M.A.; El Benna, J. Priming of human neutrophil respiratory burst by granulocyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation of p47(phox). J. Biol. Chem. 1999, 274, 20704–20708. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.; Buskiewicz-Koenig, I.A. Redox Activation of Mitochondrial DAMPs and the Metabolic Consequences for Development of Autoimmunity. Antioxid. Redox Signal. 2022, 36, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Bours, M.; Swennen, E.; Di Virgilio, F.; Cronstein, B.; Dagnelie, P. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Alula, K.; Jackson, D.; Smith, A.; Kim, D.; Turner, K.; Odstrcil, E.; Kaipparettu, B.; Dassopoulos, T.; Venuprasad, K.; Feagins, L.; et al. Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease. Cells 2021, 10, 1349. [Google Scholar] [CrossRef]
- Wang, M.; Zeng, F.; Ning, F.; Wang, Y.; Zhou, S.; He, J.; Li, C.; Wang, C.; Sun, X.; Zhang, D.; et al. Ceria nanoparticles ameliorate renal fibrosis by modulating the balance between oxidative phosphorylation and aerobic glycolysis. J. Nanobiotechnol. 2022, 20, 3. [Google Scholar] [CrossRef]
- Kovács, D.; BagónéVántus, V.; Vámos, E.; Kálmán, N.; Schicho, R.; Gallyas, F.; Radnai, B. Olaparib: A Clinically Applied PARP Inhibitor Protects from Experimental Crohn’s Disease and Maintains Barrier Integrity by Improving Bioenergetics through Rescuing Glycolysis in Colonic Epithelial Cells. Oxid. Med. Cell. Longev. 2021, 2021, 7308897. [Google Scholar] [CrossRef]
- Jurickova, I.; Bonkowski, E.; Angerman, E.; Novak, E.; Huron, A.; Akers, G.; Iwasawa, K.; Braun, T.; Hadar, R.; Hooker, M.; et al. Eicosatetraynoic Acid and Butyrate Regulate Human Intestinal Organoid Mitochondrial and Extracellular Matrix Pathways Implicated in Crohn’s Disease Strictures. Inflamm. Bowel Dis. 2022, izac037. [Google Scholar] [CrossRef]
- Ho, G.-T.; Theiss, A.L. Mitochondria and Inflammatory Bowel Diseases: Toward a Stratified Therapeutic Intervention. Annu. Rev. Physiol. 2021, 84, 435–459. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagov, A.; Zhigmitova, E.B.; Sazonova, M.A.; Mikhaleva, L.M.; Kalmykov, V.; Shakhpazyan, N.K.; Orekhova, V.A.; Orekhov, A.N. Novel Models of Crohn’s Disease Pathogenesis Associated with the Occurrence of Mitochondrial Dysfunction in Intestinal Cells. Int. J. Mol. Sci. 2022, 23, 5141. https://doi.org/10.3390/ijms23095141
Blagov A, Zhigmitova EB, Sazonova MA, Mikhaleva LM, Kalmykov V, Shakhpazyan NK, Orekhova VA, Orekhov AN. Novel Models of Crohn’s Disease Pathogenesis Associated with the Occurrence of Mitochondrial Dysfunction in Intestinal Cells. International Journal of Molecular Sciences. 2022; 23(9):5141. https://doi.org/10.3390/ijms23095141
Chicago/Turabian StyleBlagov, Alexander, Elena B. Zhigmitova, Margarita A. Sazonova, Liudmila M. Mikhaleva, Vladislav Kalmykov, Nikolay K. Shakhpazyan, Varvara A. Orekhova, and Alexander N. Orekhov. 2022. "Novel Models of Crohn’s Disease Pathogenesis Associated with the Occurrence of Mitochondrial Dysfunction in Intestinal Cells" International Journal of Molecular Sciences 23, no. 9: 5141. https://doi.org/10.3390/ijms23095141