
Cullin 3 Exon 9 Deletion in Familial Hyperkalemic Hypertension Impairs Cullin3-Ring-E3 Ligase (CRL3) Dynamic Regulation and Cycling

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Interactome Comparison of CUL3-WT and CUL3-∆9 in Stable and Inducible Cell Lines
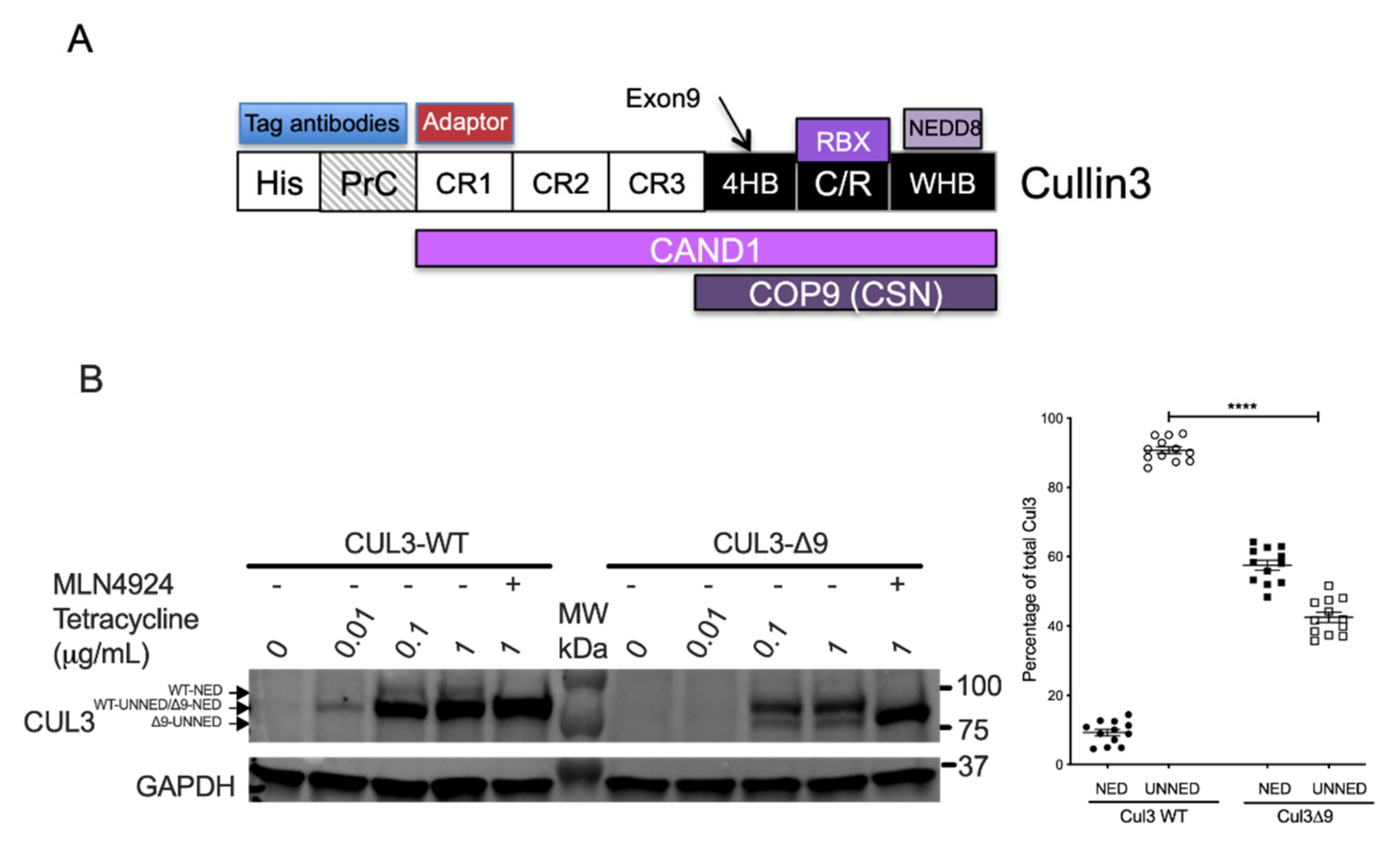
2.2. Exon 9 Deletion Does Not Modify the Dimerization of the CUL3-Rbx1 Complex
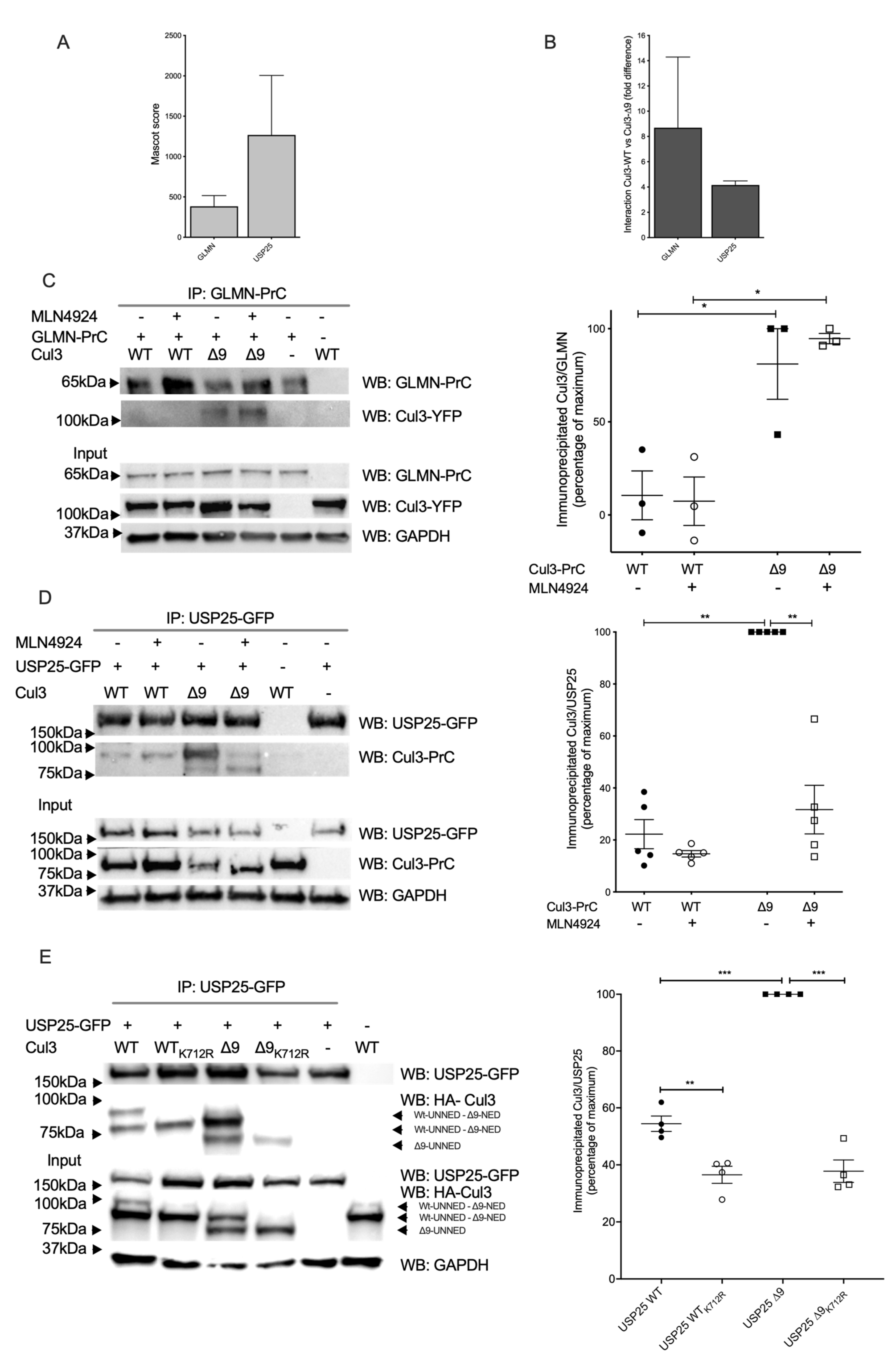
2.3. Among Several CRL3 Regulators, Glomulin, and USP25 Interact More with CUL3-∆9
2.4. COP9 Signalosome and CAND1, Two Major Regulators of CRL Cycling, Do Not Interact with CUL3-∆9
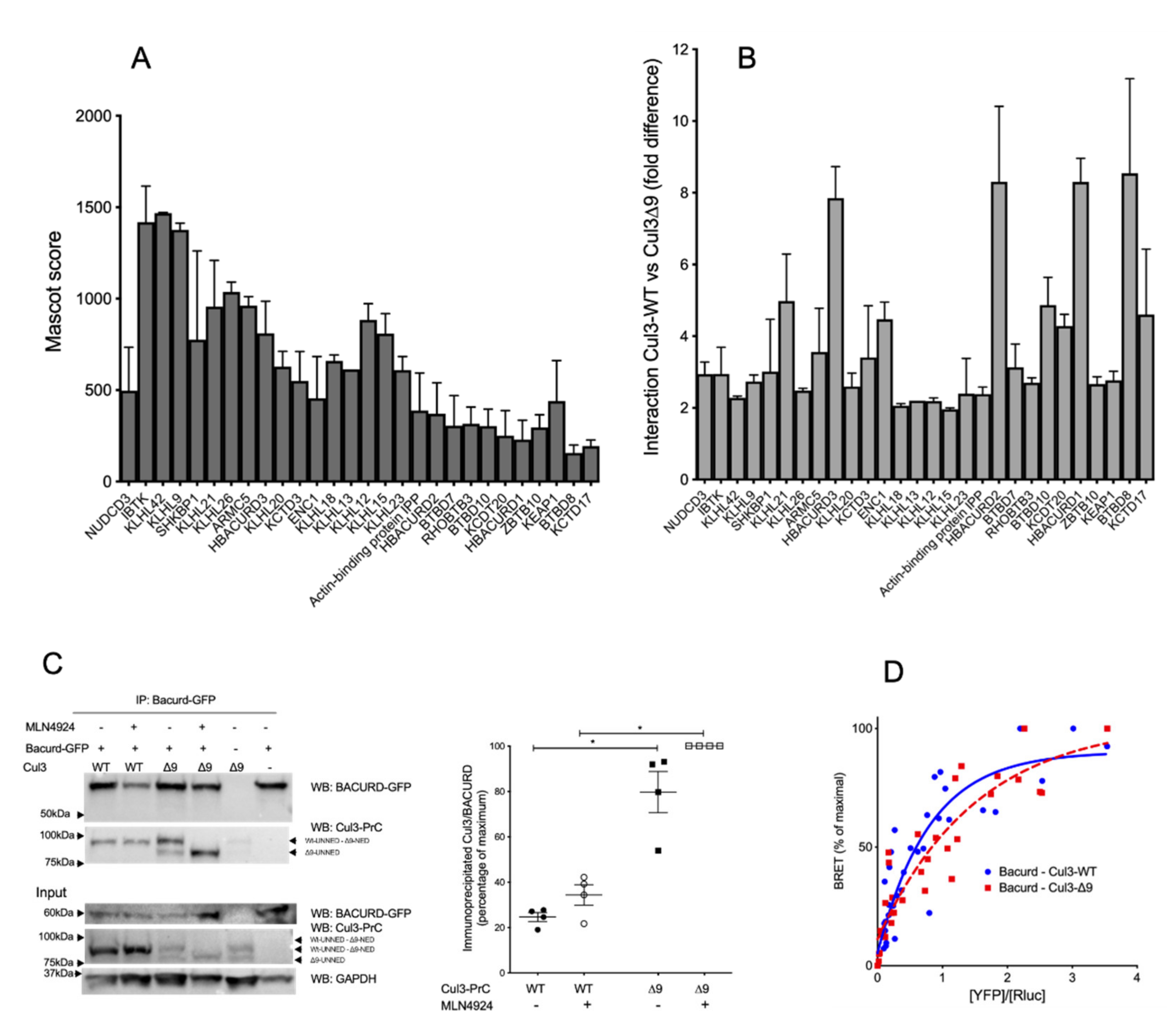
2.5. CUL3-∆9 Interacts More Efficiently with Numerous BTB-Domain Containing Adaptors
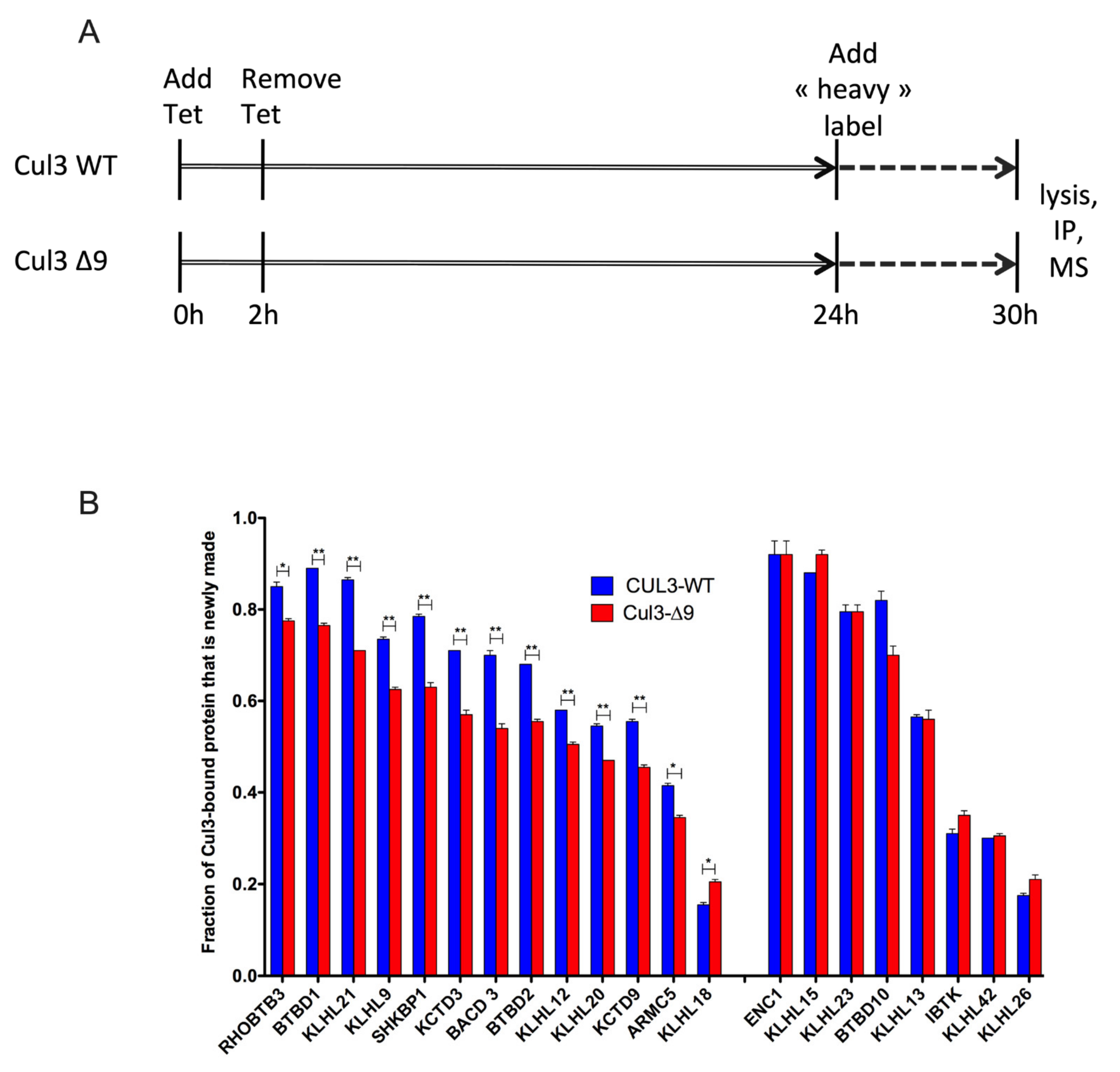
2.6. Exploring the Dynamic Nature of CUL3-∆9-Associated Proteome by SILAC-Based Quantitative Mass Spectrometry
3. Discussion
4. Materials and Methods
4.1. Vectors
4.2. Cell Culture and Transfection
4.3. Interactome Analysis Using LC-MS/MS
4.3.1. Samples Preparation
4.3.2. LC-MS/MS Acquisition
4.3.3. LC-MS/MS Data Processing and Analysis
4.4. Reciprocal Co-Immunoprecipitations of CUL3-WT and CUL3-∆9
4.4.1. Immunoprecipitation
4.4.2. Immunoblotting
4.5. Degradation and BRET Experiments
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Genschik, P.; Sumara, I.; Lechner, E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): Cellular functions and disease implications. EMBO J. 2013, 32, 2307–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cope, G.A.; Suh, G.S.; Aravind, L.; Schwarz, S.E.; Zipursky, S.L.; Koonin, E.V.; Deshaies, R.J. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 2002, 298, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Lyapina, S.; Cope, G.; Shevchenko, A.; Serino, G.; Tsuge, T.; Zhou, C.; Wolf, D.A.; Wei, N.; Shevchenko, A.; Deshaies, R.J. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science 2001, 292, 1382–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusho, T.; Uchida, S.; Sohara, E. The WNK signaling pathway and salt-sensitive hypertension. Hypertens. Res. 2020, 43, 733–743. [Google Scholar] [CrossRef]
- Glover, M.; Ware, J.S.; Henry, A.; Wolley, M.; Walsh, R.; Wain, L.V.; Xu, S.; Van’t Hoff, W.G.; Tobin, M.D.; Hall, I.P.; et al. Detection of mutations in KLHL3 and CUL3 in families with FHHt (familial hyperkalaemic hypertension or Gordon’s syndrome). Clin. Sci. 2014, 126, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Ibeawuchi, S.R.; Agbor, L.N.; Quelle, F.W.; Sigmund, C.D. Hypertension-causing mutations in Cullin3 protein impair RhoA protein ubiquitination and augment the association with substrate adaptors. J. Biol. Chem. 2015, 290, 19208–19217. [Google Scholar] [CrossRef] [Green Version]
- McCormick, J.A.; Yang, C.L.; Zhang, C.; Davidge, B.; Blankenstein, K.I.; Terker, A.S.; Yarbrough, B.; Meermeier, N.P.; Park, H.J.; McCully, B.; et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J. Clin. Investig. 2014, 124, 4723–4736. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, F.R.; Siew, K.; Zhang, J.; Johnson, C.; Wood, N.; Cleary, S.E.; Al Maskari, R.S.; Ferryman, J.T.; Hardege, I.; Figg, N.; et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol. Med. 2015, 7, 1285–1306. [Google Scholar] [CrossRef]
- Wimuttisuk, W.; Singer, J.D. The Cullin3 ubiquitin ligase functions as a Nedd8-bound heterodimer. Mol. Biol. Cell 2007, 18, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achour, L.; Kamal, M.; Jockers, R.; Marullo, S. Using quantitative BRET to assess G protein-coupled receptor homo- and heterodimerization. Methods Mol. Biol. 2011, 756, 183–200. [Google Scholar] [PubMed]
- Brouillard, P.; Boon, L.M.; Mulliken, J.B.; Enjolras, O.; Ghassibe, M.; Warman, M.L.; Tan, O.T.; Olsen, B.R.; Vikkula, M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am. J. Hum. Genet. 2002, 70, 866–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.M.; Olszewski, J.L.; Tron, A.E.; Hammel, M.; Lambert, L.J.; Waddell, M.B.; Mittag, T.; DeCaprio, J.A.; Schulman, B.A. Structure of a glomulin-RBX1-CUL1 complex: Inhibition of a RING E3 ligase through masking of its E2-binding surface. Mol. Cell 2012, 47, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Comas, A.; Lindsten, K.; Gonzalez-Duarte, R.; Masucci, M.G.; Marfany, G. The ubiquitin-specific protease USP25 interacts with three sarcomeric proteins. Cell Mol. Life Sci. 2006, 63, 723–734. [Google Scholar] [CrossRef]
- Goldenberg, S.J.; Cascio, T.C.; Shumway, S.D.; Garbutt, K.C.; Liu, J.; Xiong, Y.; Zheng, N. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell 2004, 119, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 2013, 153, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhu, W.; Nhan, T.; Toth, J.I.; Petroski, M.D.; Wolf, D.A. CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nat. Commun. 2013, 4, 1642. [Google Scholar] [CrossRef]
- Zemla, A.; Thomas, Y.; Kedziora, S.; Knebel, A.; Wood, N.T.; Rabut, G.; Kurz, T. CSN- and CAND1-dependent remodelling of the budding yeast SCF complex. Nat. Commun. 2013, 4, 1641. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef]
- Abdel Khalek, W.; Rafael, C.; Loisel-Ferreira, I.; Kouranti, I.; Clauser, E.; Hadchouel, J.; Jeunemaitre, X. Severe arterial hypertension from Cullin 3 mutations is caused by both renal and vascular effects. J. Am. Soc. Nephrol. 2019, 30, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, W.; Du, Y.; Li, Z.; Xuan, Z.; Zhao, L.; Chen, J.; Zhao, Y.; Tuo, B.; Zheng, S.; et al. Inhibition of KLHL21 prevents cholangiocarcinoma progression through regulating cell proliferation and motility, arresting cell cycle and reducing Erk activation. Biochem. Biophys. Res. Commun. 2018, 499, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Courtheoux, T.; Enchev, R.I.; Lampert, F.; Gerez, J.; Beck, J.; Picotti, P.; Sumara, I.; Peter, M. Cortical dynamics during cell motility are regulated by CRL3(KLHL21) E3 ubiquitin ligase. Nat. Commun. 2016, 7, 12810. [Google Scholar]
- Friedman, J.S.; Ray, J.W.; Waseem, N.; Johnson, K.; Brooks, M.J.; Hugosson, T.; Breuer, D.; Branham, K.E.; Krauth, D.S.; Bowne, S.J.; et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 2009, 84, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kigoshi, Y.; Tsuruta, F.; Chiba, T. Ubiquitin ligase activity of Cul3-KLHL7 protein is attenuated by autosomal dominant retinitis pigmentosa causative mutation. J. Biol. Chem. 2011, 286, 33613–33621. [Google Scholar] [CrossRef] [Green Version]
- Maerki, S.; Olma, M.H.; Staubli, T.; Steigemann, P.; Gerlich, D.W.; Quadroni, M.; Sumara, I.; Peter, M. The Cul3-KLHL21 E3 ubiquitin ligase targets aurora B to midzone microtubules in anaphase and is required for cytokinesis. J. Cell. Biol. 2009, 187, 791–800. [Google Scholar] [CrossRef]
- Nawa, M.; Kanekura, K.; Hashimoto, Y.; Aiso, S.; Matsuoka, M. A novel Akt/PKB-interacting protein promotes cell adhesion and inhibits familial amyotrophic lateral sclerosis-linked mutant SOD1-induced neuronal death via inhibition of PP2A-mediated dephosphorylation of Akt/PKB. Cell Signal. 2008, 20, 493–505. [Google Scholar] [CrossRef]
- Nawa, M.; Matsuoka, M. KCTD20, a relative of BTBD10, is a positive regulator of Akt. BMC Biochem. 2013, 14, 27. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Yao, S.; Jing, L.; He, J.; Xiao, C.; Zhang, T.; Meng, W.; Zhu, H.; Xu, H.; Mo, X. ENC1-like integrates the retinoic acid/FGF signaling pathways to modulate ciliogenesis of Kupffer’s Vesicle during zebrafish embryonic development. Dev. Biol. 2013, 374, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, R.J.; Zhang, C.; Erspamer, K.J.; Agbor, L.N.; Sigmund, C.D.; Singer, J.D.; Yang, C.L.; Ellison, D.H. Dual gain and loss of cullin 3 function mediates familial hyperkalemic hypertension. Am. J. Physiol. Renal. Physiol. 2018, 315, F1006–F1018. [Google Scholar] [CrossRef]
- Ferdaus, M.Z.; Miller, L.N.; Agbor, L.N.; Saritas, T.; Singer, J.D.; Sigmund, C.D.; McCormick, J.A. Mutant Cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight 2017, 2, e96700. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Sorrell, F.J.; Alessi, D.R.; Bullock, A.N.; Kurz, T. Structural and biochemical characterization of the KLHL3-WNK kinase interaction important in blood pressure regulation. Biochem. J. 2014, 460, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, M.; Mori, T.; Isobe, K.; Sohara, E.; Susa, K.; Araki, Y.; Chiga, M.; Kikuchi, E.; Nomura, N.; Mori, Y.; et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013, 3, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Araki, Y.; Mori, T.; Sasaki, E.; Kasagi, Y.; Isobe, K.; Susa, K.; Inoue, Y.; Bomont, P.; Okado, T.; et al. Decreased KLHL3 expression is involved in the pathogenesis of pseudohypoaldosteronism type II caused by cullin 3 mutation in vivo. Clin. Exp. Nephrol. 2018, 22, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Wimuttisuk, W.; West, M.; Davidge, B.; Yu, K.; Salomon, A.; Singer, J.D. Novel Cul3 binding proteins function to remodel E3 ligase complexes. BMC Cell Biol. 2014, 15, 28. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wei, Y.; Reboul, J.; Vaglio, P.; Shin, T.H.; Vidal, M.; Elledge, S.J.; Harper, J.W. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 2003, 425, 316–321. [Google Scholar] [CrossRef]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [Green Version]
- Chew, E.H.; Hagen, T. Substrate-mediated regulation of cullin neddylation. J. Biol. Chem. 2007, 282, 17032–17040. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, R.J.; Yang, C.L.; Ellison, D.H. Hypertension-causing cullin 3 mutations disrupt COP9 signalosome binding. Am. J. Physiol. Renal. Physiol. 2020, 318, F204–F208. [Google Scholar] [CrossRef]
- Araki, Y.; Rai, T.; Sohara, E.; Mori, T.; Inoue, Y.; Isobe, K.; Kikuchi, E.; Ohta, A.; Sasaki, S.; Uchida, S. Generation and analysis of knock-in mice carrying pseudohypoaldosteronism type II-causing mutations in the cullin 3 gene. Biol. Open 2015, 4, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Canales, M.; Zhang, C.; Soukaseum, C.; Moreno, E.; Pacheco-Alvarez, D.; Vidal-Petiot, E.; Castaneda-Bueno, M.; Vazquez, N.; Rojas-Vega, L.; Meermeier, N.P.; et al. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 2014, 64, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olma, M.H.; Roy, M.; Le Bihan, T.; Sumara, I.; Maerki, S.; Larsen, B.; Quadroni, M.; Peter, M.; Tyers, M.; Pintard, L. An interaction network of the mammalian COP9 signalosome identifies Dda1 as a core subunit of multiple Cul4-based E3 ligases. J. Cell Sci. 2009, 122, 1035–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derivery, E.; Gautreau, A. Assaying WAVE and WASH complex constitutive activities toward the Arp2/3 complex. Methods Enzymol. 2010, 484, 677–695. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Score | Anova | Fold | |
|---|---|---|---|---|
| IBTK | 32/26 | 1615/1221 | 0.02/0.01 | 2.2/3.69 |
| KBTBD6 | 31/ | 1585/ | 0.05/ | 1.39/ |
| KLHL42 | 26/22 | 1462/1472 | 0.000709/0.02 | 2.23/2.33 |
| KLHL9 | 29/23 | 1413/1340 | 0.01/0.00825 | 2.92/2.55 |
| SHKBP1 | 23/6 | 1261/289 | 0.000149/0.11 | 4.47/1.55 |
| KLHL22 | 24/15 | 1238/770 | 0.03/0.65 | 1.67/1.06 |
| KLHL21 | 21/13 | 1209/704 | 0.03/0.01 | 6.29/3.67 |
| KLHL26 | 18/14 | 1091/983 | 0.0047/0.00809 | 2.42/2.55 |
| KLHL8 | 21/11 | 1041/582 | 0.43/0.06 | 1.14/1.36 |
| KCTD9 | 13/ | 1024/ | 0.13/ | 2.11/ |
| BACD3 | 16/12 | 986/635 | 0.02/0.05 | 6.97/8.73 |
| KLHL15 | 16/13 | 918/700 | 0.00887/0.03 | 1.92/2 |
| BTBD9 | 18/11 | 886/524 | 0.38/0.19 | 1.08/1.72 |
| KLHL12 | 13/17 | 796/972 | 0.07/0.0065 | 2.1/2.28 |
| KLHL20 | 15/12 | 712/545 | 0.02/0.01 | 2.22/2.97 |
| KCTD3 | 14/7 | 711/389 | 0.01/0.02 | 4.85/1.96 |
| KLHL23 | 14/9 | 684/535 | 0.13/0.01 | 1.41/3.38 |
| ENC1 | 13/3 | 683/228 | 0.00265/0.03 | 3.99/4.95 |
| KLHL7 | 13/3 | 649/168 | 0.07/0.19 | 1.31/1.34 |
| BTBD2 | 8/11 | 627/742 | 0.19/0.15 | 1.8/2.58 |
| KLHL18 | 12/9 | 627/693 | 0.01/0.36 | 2/2.12 |
| KLHL13 | 12/ | 614/ | 0.02/ | 2.2/ |
| ACTIN-BINDING PROTEIN IPP | 12/4 | 594/183 | 0.05/0.000248 | 2.19/2.58 |
| KBTBD7 | 10/21 | 553/1281 | 0.1/0.36 | 1.37/1.09 |
| BACD2 | 8/6 | 540/201 | 0.02/0.000164 | 10.41/6.2 |
| RCBTB1 | 9/5 | 524/214 | 0.83/0.14 | 1.13/1.1 |
| RHOBTB1 | 12/4 | 521/194 | 0.2/0.32 | 1.78/1.31 |
| KBTBD4 | 8/5 | 501/208 | 0.1/0.67 | 1.43/1.77 |
| BTBD7 | 11/2 | 470/139 | 0.03/0.09 | 3.78/2.48 |
| KLHL36 | 9/2 | 455/97 | 0.18/0.49 | 1.15/2.28 |
| KLHL25 | 10/5 | 453/318 | 0.37/0.00372 | 1.31/1.4 |
| RHOBTB3 | 10/4 | 407/224 | 0.000957/0.1 | 2.84/2.56 |
| KBTBD2 | 7/ | 402/ | 0.63/ | 1.14/ |
| BTBD10 | 6/5 | 396/211 | 0.04/0.01 | 5.64/4.09 |
| KCTD20 | 11/4 | 389/112 | 0.02/0.00129 | 3.95/4.61 |
| KLHL17 | 10/ | 376/ | 0.38/ | 1.17/ |
| KBTBD8 | 10/ | 369/ | 0.00828/ | 4.39/ |
| BACD1 | 5/4 | 335/123 | 0.00486/0.000495 | 8.96/7.64 |
| KCTD18 | 7/9 | 302/312 | 0.03/0.58 | 1.44/1.09 |
| RCBTB2 | 5/3 | 259/115 | 0.65/0.03 | 1.21/1.56 |
| GIGAXONIN | 7/ | 250/ | 0.09/ | 1.58/ |
| BTBD1 | 5/9 | 229/558 | 0.32/0.06 | 2.73/2.66 |
| ZBTB10 | 6/8 | 226/365 | 0.01/0.000536 | 2.87/2.46 |
| KEAP1 | 5/11 | 219/661 | 0.02/0.00048 | 3.02/2.51 |
| KLHL24 | 5/4 | 206/178 | 0.34/0.59 | 1.44/1.26 |
| BTBD8 | 5/3 | 200/112 | 0.03/0.00296 | 5.9/11.18 |
| KCTD6 | 4/ | 182/ | 0.64/ | 1.29/ |
| KLHL28 | 5/2 | 172/123 | 0.2/0.71 | 1.3/1.19 |
| KCTD17 | 5/4 | 159/228 | 0.02/0.16 | 6.43/2.77 |
| KCTD2 | 2/2 | 118/156 | 0.08/0.21 | 6.44/3.19 |
| ZNF131 | 3/ | 104/ | 0.59/ | 2.21/ |
| KLHL11 | 3/ | 100/ | 0.25/ | 1.33/ |
| KLHL5 | 4/2 | 82/136 | 0.24/0.17 | 1.49/1.69 |
| BTBD6 | 2/ | 76/ | 0.02/ | 8.91/ |
| KLHL2 | 2/ | 71/ | 0.76/ | 1.19/ |
| ZBTB7A | /4 | /197 | /0.33 | /2.21 |
| KCTD5 | /2 | /98 | /0.23 | /2.75 |
| BACH2 | /11 | /538 | /0.00639 | /2.5 |
| Galectin-3-binding protein | /4 | /143 | /0.04 | /2.44 |
| KAISO | /3 | /127 | /0.04 | /3.13 |
| BACH1 | /15 | /840 | /0.01 | /2.63 |
| BTBD11 | /10 | /645 | /0.01 | /1.95 |
| ZBTB14 | /4 | /216 | /0.11 | /2.95 |
| ZBTB21 | /3 | /83 | /0.17 | /1.82 |
| ZBTB1 | /2 | /51 | /0.12 | /2.3 |
| ZBTB17 | /2 | /56 | /0.43 | /1.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouranti, I.; Abdel Khalek, W.; Mazurkiewicz, S.; Loisel-Ferreira, I.; Gautreau, A.M.; Pintard, L.; Jeunemaitre, X.; Clauser, E. Cullin 3 Exon 9 Deletion in Familial Hyperkalemic Hypertension Impairs Cullin3-Ring-E3 Ligase (CRL3) Dynamic Regulation and Cycling. Int. J. Mol. Sci. 2022, 23, 5151. https://doi.org/10.3390/ijms23095151
Kouranti I, Abdel Khalek W, Mazurkiewicz S, Loisel-Ferreira I, Gautreau AM, Pintard L, Jeunemaitre X, Clauser E. Cullin 3 Exon 9 Deletion in Familial Hyperkalemic Hypertension Impairs Cullin3-Ring-E3 Ligase (CRL3) Dynamic Regulation and Cycling. International Journal of Molecular Sciences. 2022; 23(9):5151. https://doi.org/10.3390/ijms23095151
Chicago/Turabian StyleKouranti, Ilektra, Waed Abdel Khalek, Stephani Mazurkiewicz, Irmine Loisel-Ferreira, Alexis M. Gautreau, Lionel Pintard, Xavier Jeunemaitre, and Eric Clauser. 2022. "Cullin 3 Exon 9 Deletion in Familial Hyperkalemic Hypertension Impairs Cullin3-Ring-E3 Ligase (CRL3) Dynamic Regulation and Cycling" International Journal of Molecular Sciences 23, no. 9: 5151. https://doi.org/10.3390/ijms23095151