
Mechanisms of Resistance to Spot Blotch in Yunnan Iron Shell Wheat Based on Metabolome and Transcriptomics

Abstract
:1. Introduction
2. Results
2.1. Detection Results and Analysis of Widely Targeted Metabolome
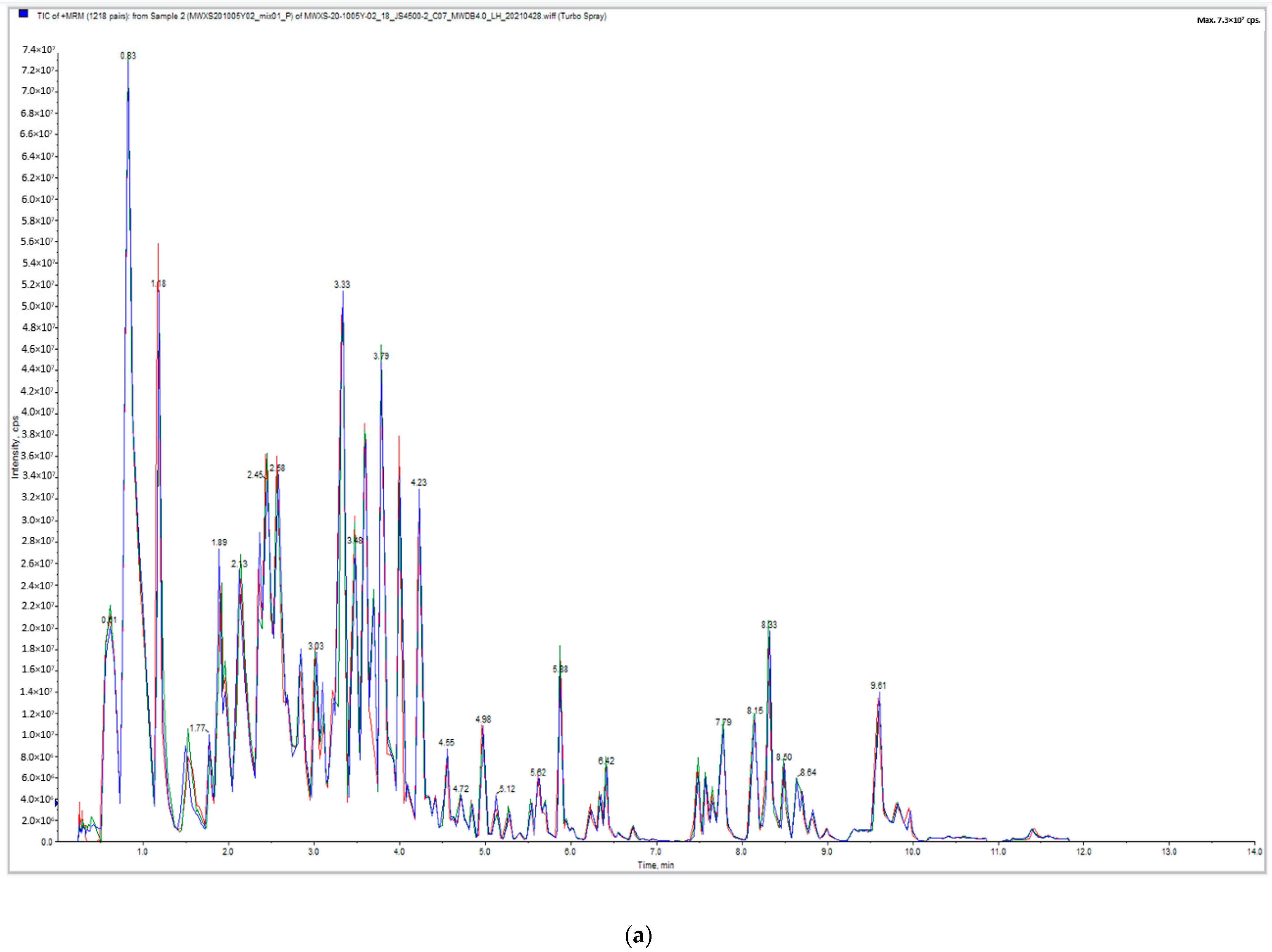
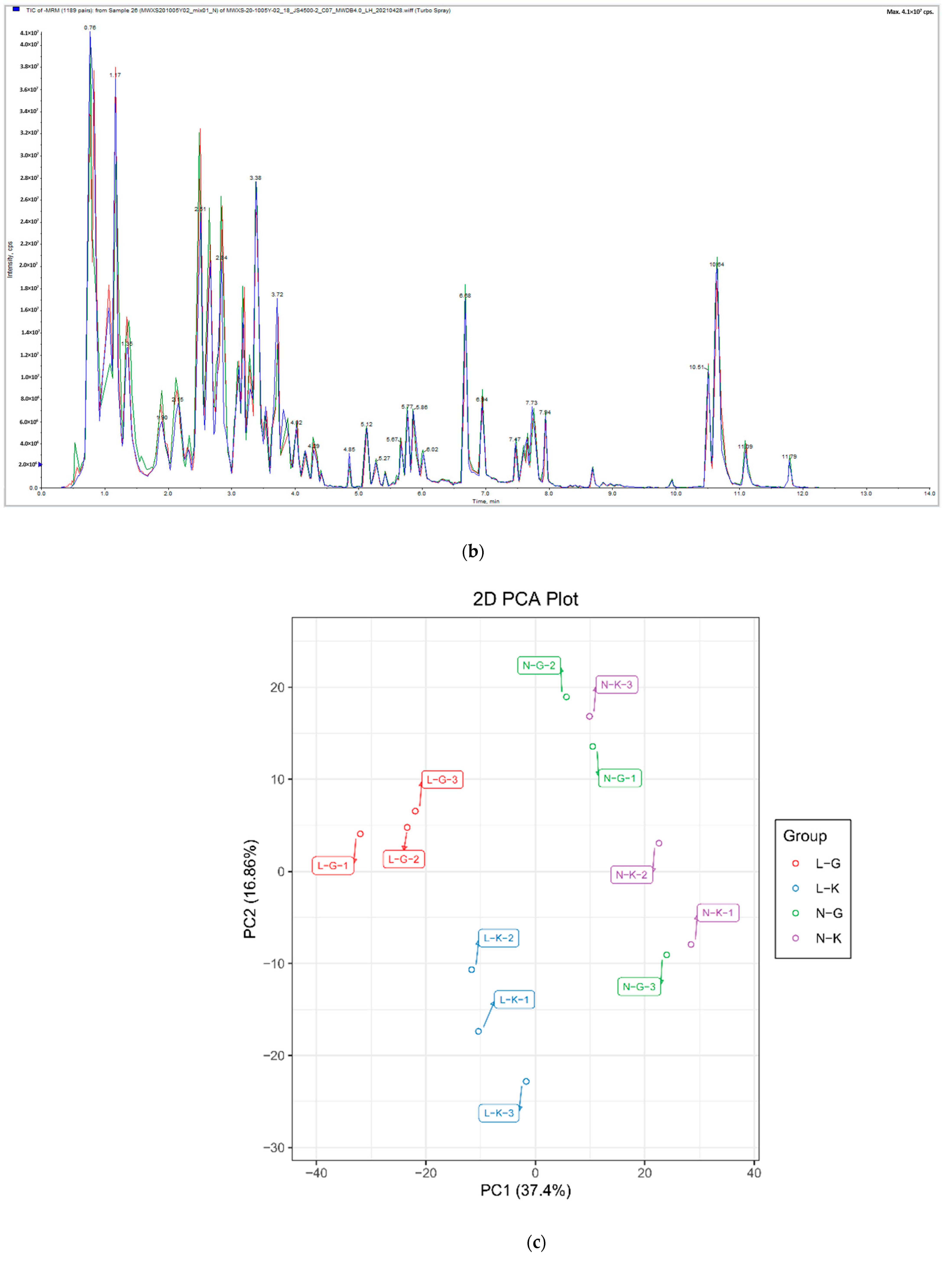
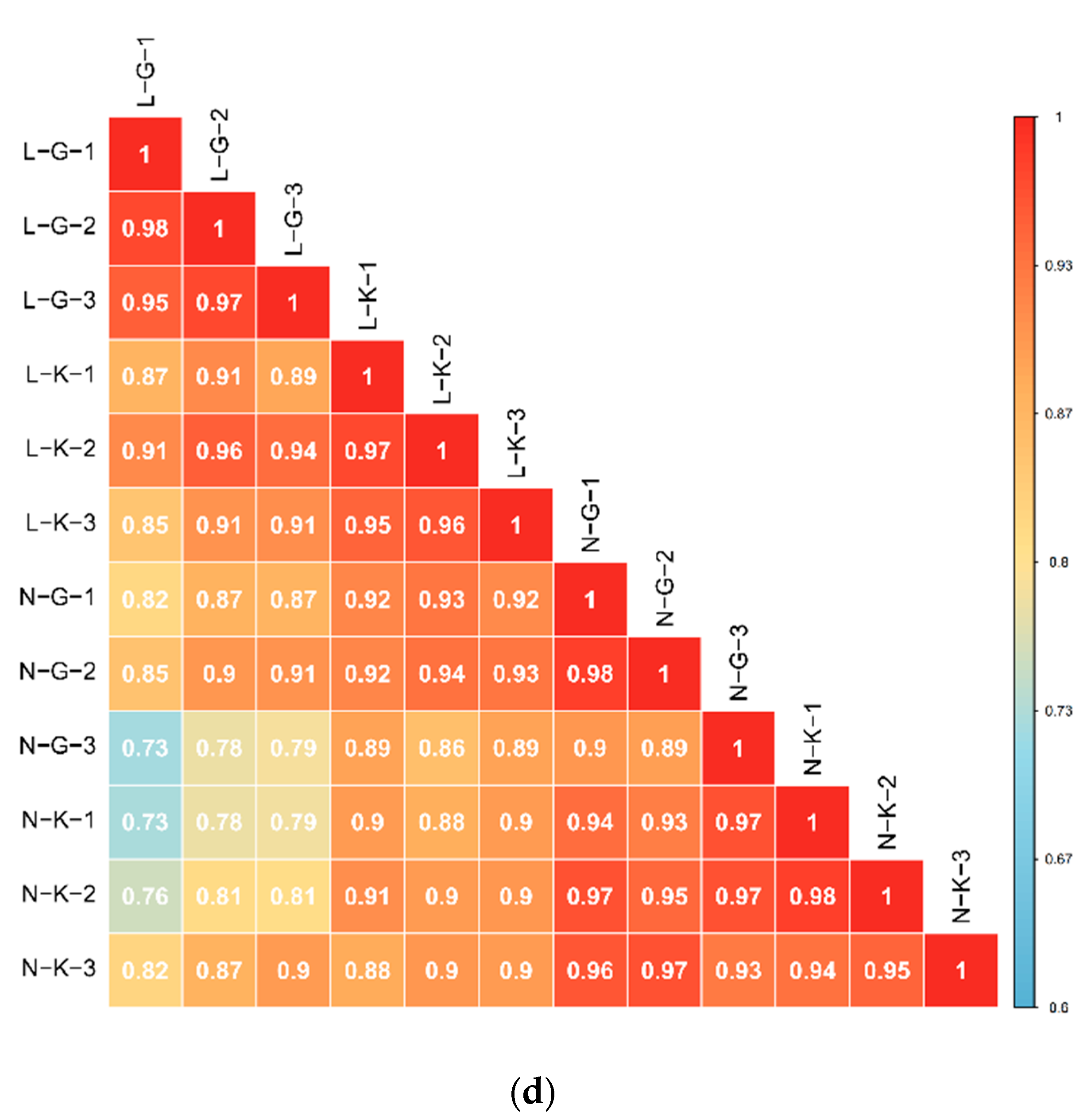
2.1.1. Quality Control of Widely Targeted Metabolome Data
2.1.2. Qualitative and Quantitative Analysis of Metabolome
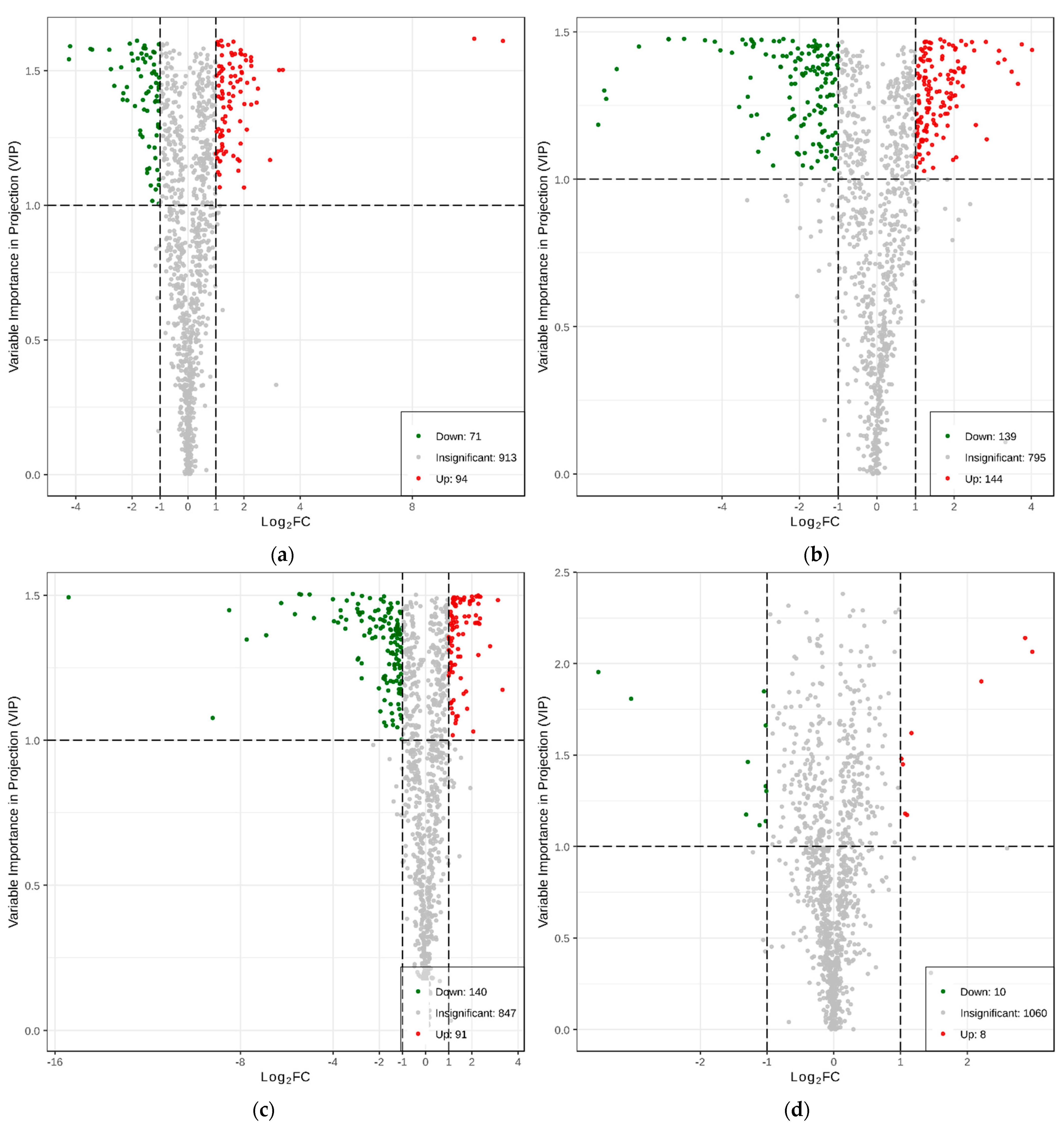
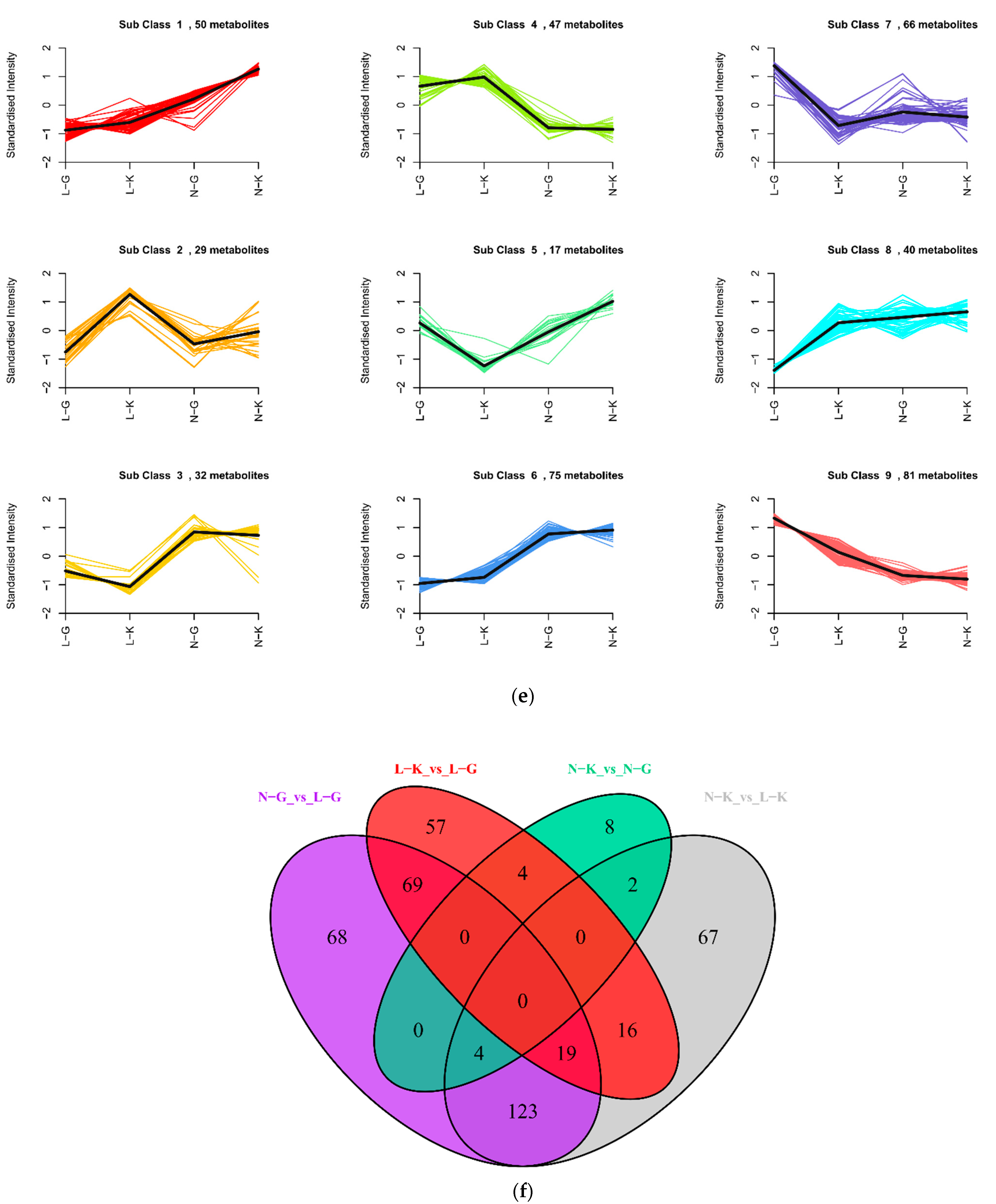
2.1.3. Differential Analysis of Metabolic Profiles between Resistant and Susceptible Strains
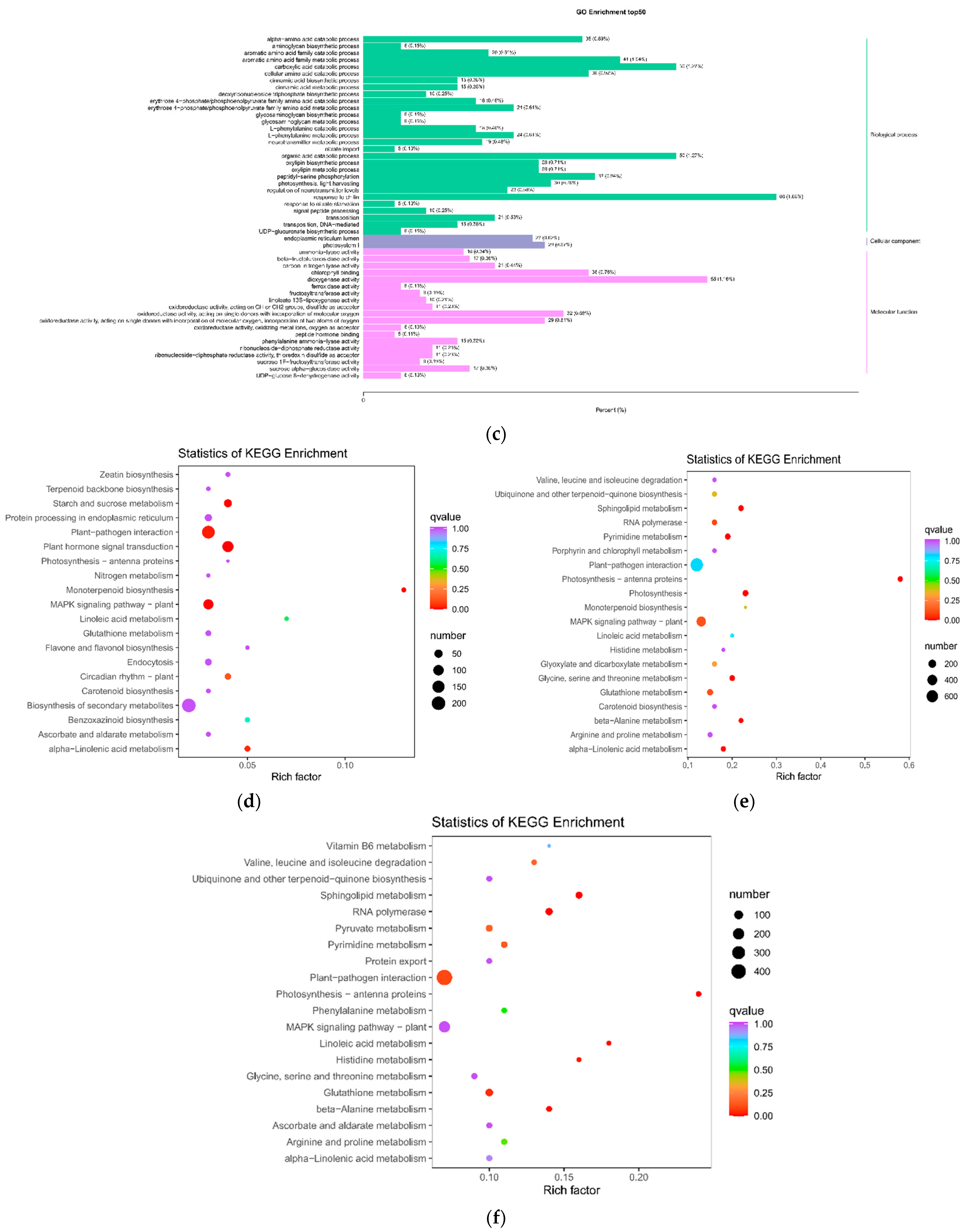
2.1.4. Enrichment Analysis of Differential Metabolite KEGG Pathway after SB Infection
2.2. Transcriptome Test Results and Analysis
2.2.1. Assembly Notes
2.2.2. Differential Genes and GO and KEGG Enrichment Analysis
2.2.3. Screening of Genes Related to Metabolic Pathway Differences
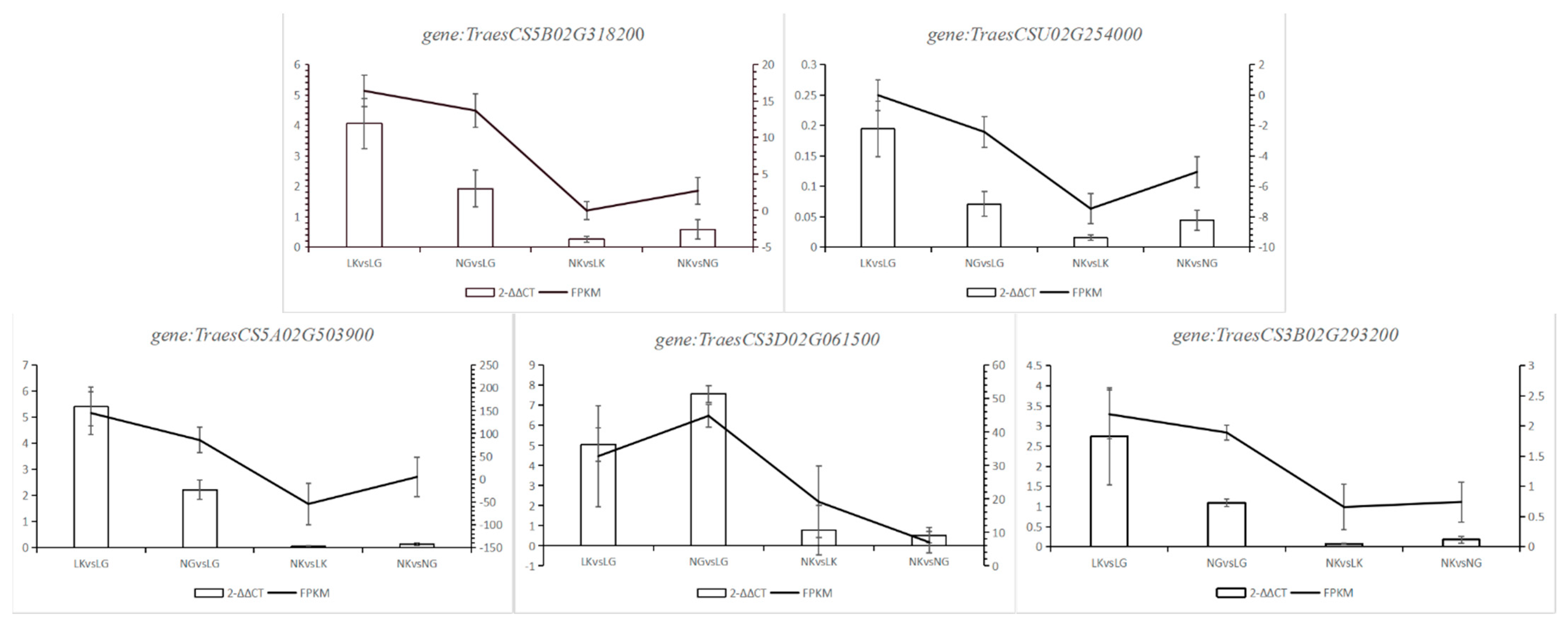
2.2.4. RT-qPCR Validation
2.3. Combined Analysis of Transcription and Metabolome
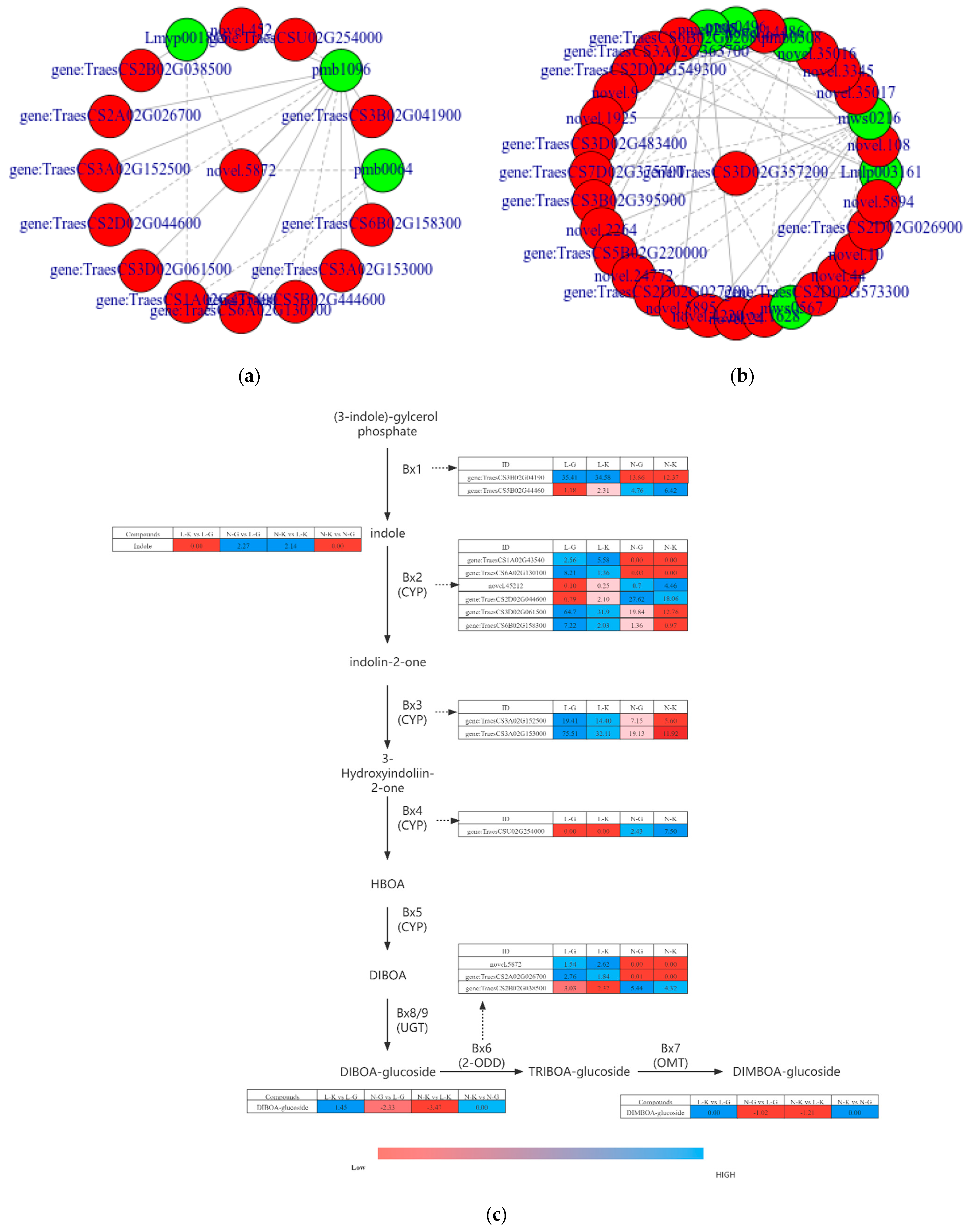
2.3.1. Connection between DEGs and Dams
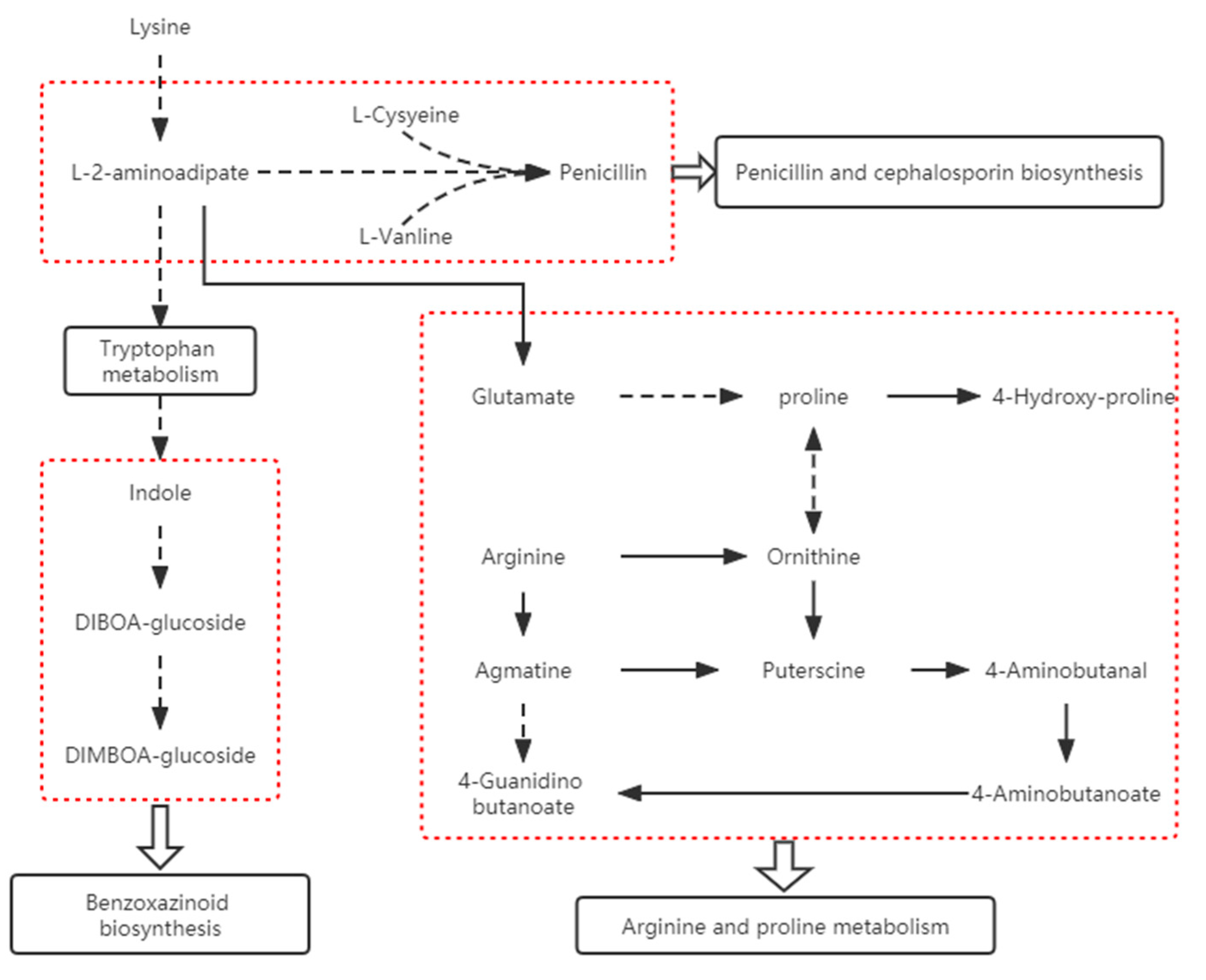
2.3.2. Correlation Analysis of Differential Genes and Metabolites in “Benzoxazinoid Biosynthesis” and “Arginine and Proline Metabolism”
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Widely Targeted Metabolome Detection and Analysis
4.3. Transcriptome Sequencing and Analysis
4.4. Combined Analysis of Transcriptome and Metabolome
4.5. RT-qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Dong, L.; Hu, J.; Pang, Y.; Hu, L.; Xiao, G.; Ma, X.; Kong, X.; Jia, J.; Wang, H.; et al. Dissecting genetic loci affecting grain morphological traits to improve grain weight via nested association mapping. Theor. Appl. Genet. 2019, 132, 3115–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jiang, Y.; Yao, F.; Long, L.; Wang, Y.; Wu, Y.; Li, H.; Wang, J.; Jiang, Q.; Kang, H.; et al. Genome-Wide Association Study Reveals the Genetic Architecture of Stripe Rust Resistance at the Adult Plant Stage in Chinese Endemic Wheat. Front. Plant Sci. 2020, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zheng, D.; Qiao, D.; Zeng, X.; En, Z.; Chen, X. Expedition and investigation of Yunnan wheat (Triticum aestivum ssp. yunnanense King). Acta Agronom. Sin. 1981, 7, 145–152. (In Chinese) [Google Scholar]
- Dong, Y.S.; Zheng, D.S. Chinese Wheat Genetic Resources; China Agriculture Press: Beijing, China, 1999; pp. 53–56. [Google Scholar]
- Zhang, X.Y.; Pang, B.S.; You, G.X.; Wang, L.F.; Jia, J.Z.; Dong, Y.C. Allelic Variation and Genetic Diversity at Glu-1 Loci in Chinese Wheat (Triticum aestivum L.) Germplasms. Agric. Sci. China 2002, 1, 1074–1082. [Google Scholar]
- Ayana, G.T.; Ali, S.; Sidhu, J.S.; Gonzalez Hernandez, J.L.; Turnipseed, B.; Sehgal, S.K. Genome-Wide Association Study for Spot Blotch Resistance in Hard Winter Wheat. Front. Plant Sci. 2018, 9, 926. [Google Scholar] [CrossRef]
- Hagerty, C.H.; Graebner, R.C.; Sackett, K.E.; Mundt, C.C. Variable competitive effects of fungicide resistance in field experiments with a plant pathogenic fungus. Ecol. Appl. 2017, 27, 1305–1316. [Google Scholar] [CrossRef]
- Eriksen, L.; Shaw, M.W.; Østergård, H. A Model of the Effect of Pseudothecia on Genetic Recombination and Epidemic Development in Populations of Mycosphaerella graminicola. Phytopathology 2001, 91, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Kema, G.H.; Verstappen, E.C.; Todorova, M.; Waalwijk, C. Successful crosses and molecular tetrad and progeny analyses demonstrate heterothallism in Mycosphaerella graminicola. Curr. Genet. 1996, 30, 251–258. [Google Scholar] [CrossRef]
- Joshi, A.K.; Chand, R. Variation and inheritance of leaf angle, and its association with spot blotch (Bipolaris sorokiniana) severity in wheat (Triticum aestivum). Euphytica 2002, 124, 283–291. [Google Scholar] [CrossRef]
- Joshi, A.K.; Ortiz-Ferrara, G.; Crossa, J.; Singh, G.; Alvarado, G.; Bhatta, M.R.; Duveiller, E.; Sharma, R.C.; Pandit, D.B.; Siddique, A.B.; et al. Associations of environments in South Asia based on spot blotch disease of wheat caused by Bipolaris sorokiniana. Crop Sci. 2007, 47, 1071–1081. [Google Scholar] [CrossRef]
- Luo, C.; Ma, L.; Zhu, J.; Guo, Z.; Dong, K.; Dong, Y. Effects of Nitrogen and Intercropping on the Occurrence of Wheat Powdery Mildew and Stripe Rust and the Relationship with Crop Yield. Front. Plant Sci. 2021, 12, 637393. [Google Scholar] [CrossRef] [PubMed]
- Manning, V.A.; Hardison, L.K.; Ciuffetti, L.M. Ptr ToxA interacts with a chloroplast-localized protein. Mol. Plant-Microbe Interact. 2007, 20, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxesena, R.R.; Mishra, V.K.; Chand, R.; Chowdhury, A.K.; Bhattacharya, P.M.; Joshi, A.K. Pooling together spot blotch resistance, high yield with earliness in wheat for eastern Gangetic plains of south Asia. Field Crop. Res. 2017, 214, 291–300. [Google Scholar] [CrossRef]
- Tomar, V.; Singh, D.; Dhillon, G.S.; Singh, R.P.; Poland, J.; Joshi, A.K.; Singh, P.K.; Bhati, P.K.; Suneel Kumar, S.; Rahman, M.; et al. New QTLs for Spot Blotch Disease Resistance in Wheat (Triticum aestivum L.) Using Genome-Wide Association Mapping. Front Genet. 2021, 11, 613217. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, A.M. Bipolaris sorokiniana-Induced Black Point, Common Root Rot, and Spot Blotch Diseases of Wheat: A Review. Front Cell Infect. Microbiol. 2021, 11, 584899. [Google Scholar] [CrossRef]
- Eyal, Z.; Scharen, A.L.; Prescott, J.M.; van Ginkel, M. The Septoria Diseases of Wheat, Concepts and Methods of Disease Management; Cimmyt: Mexico City, Mexico, 1987. [Google Scholar]
- Sharma, S.; Sahu, R.; Navathe, S.; Mishra, V.K.; Chand, R.; Singh, P.K.; Joshi, A.K.; Pandey, S.P. Natural Variation in Elicitation of Defense-Signaling Associates to Field Resistance Against the Spot Blotch Disease in Bread Wheat (Triticum aestivum L.). Front Plant Sci. 2018, 9, 636. [Google Scholar] [CrossRef]
- Torriani, S.F.; Melichar, J.P.; Mills, C.; Pain, N.; Sierotzki, H.; Courbot, M. Zymoseptoria tritici: A major threat to wheat production, integrated approaches to control. Fungal Genet. Biol. 2015, 79, 8–12. [Google Scholar] [CrossRef]
- Goel, L.B.; Nagarajan, S.; Singh, R.V.; Sinha, V.C.; Kumar, J. Foliar blights of wheat: Current status in India and identification of donor lines for resistance through multilocational evaluation. Indian Phytopathol. 1999, 52, 398–402. [Google Scholar]
- Murray, G.M.; Brennan, J.P. Estimating disease losses to the Aus-tralian wheat industry. Australas Plant Path 2009, 38, 558–570. [Google Scholar] [CrossRef]
- Fones, H.; Gurr, S. The impact of Septoria tritici Blotch disease on wheat: An EU perspective. Fungal Genet. Biol. 2015, 79, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Blake, J.J.; Gosling, P.; Fraaije, B.A.; Burnett, F.J.; Knight, S.M.; Kildea, S.; Paveley, N.D. Changes in field dose–response curves for demethylation inhibitor (DMI) and quinone outside inhibitor (QoI) fungicides against Zymoseptoria tritici, related to labora-tory sensitivity phenotyping and genotyping assays. Pest Manag. Sci. 2018, 74, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Bent, A.F. Plant disease resistance genes: Function meets structure. Plant Cell 1996, 8, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Duba, A.; Goriewa-Duba, K.; Wachowska, U. A Review of the Interactions between Wheat and Wheat Pathogens: Zymoseptoria tritici, Fusarium spp. and Parastagonospora nodorum. Int. J. Mol. Sci. 2018, 19, 1138. [Google Scholar] [CrossRef] [Green Version]
- Seybold, H.; Demetrowitsch, T.J.; Hassani, M.; Szymczak, S.; Reim, E.; Haueisen, J.; Lübbers, L.; Rühlemann, M.; Franke, A.; Schwarz, K.; et al. A fungal pathogen induces systemic susceptibility and systemic shifts in wheat metabolome and microbiome composition. Nat. Commun. 2020, 11, 1910. [Google Scholar] [CrossRef] [Green Version]
- Cowley, T.; Walters, D.R. Polyamine metabolism in barley reacting hypersensitively to the powdery mildew fungus Blumeria graminis f. sp. Hordei. Plant Cell Environ. 2008, 25, 461–468. [Google Scholar] [CrossRef]
- Kaur, S.; Suseela, V. Unraveling Arbuscular Mycorrhiza-Induced Changes in Plant Primary and Secondary Metabolome. Metabolites 2020, 10, 335. [Google Scholar] [CrossRef]
- Zhu, Z.; Bonnett, D.; Ellis, M.; Singh, P.; Heslot, N.; Dreisigacker, S.; Gao, C.; Mujeeb-Kazi, A. Mapping resistance to spot blotch in a CIMMYT synthetic-derived bread wheat. Mol. Breed. 2014, 34, 1215–1228. [Google Scholar] [CrossRef]
- Lillemo, M.; Joshi, A.K.; Prasad, R.; Chand, R.; Singh, R.P. QTL for spot blotch resistance in bread wheat line Saar co-locate to the biotrophic disease resistance loci Lr34 and Lr46. Theor. Appl. Genet. 2013, 126, 711–719. [Google Scholar] [CrossRef]
- Kumar, S.; Röder, M.S.; Tripathi, S.B.; Kumar, S.; Chand, R.; Joshi, A.K.; Kumar, U. Mendelization and fne mapping of a bread wheat spot blotch disease resistance QTL. Mol. Breed 2015, 35, 218. [Google Scholar] [CrossRef]
- Lu, P.; Liang, Y.; Li, D.; Wang, Z.; Li, W.; Wang, G.; Wang, Y.; Zhou, S.H.; Wu, Q.H.; Xie, J.Z.; et al. Fine genetic mapping of spot blotch resistance gene Sb3 in wheat (Triticum aestivum). Theor. Appl. Genet. 2016, 129, 577–589. [Google Scholar] [CrossRef]
- Zhang, P.; Guo, G.; Wu, Q.; Chen, Y.; Xie, J.; Lu, P.; Li, B.; Dong, L.; Li, M.; Wang, R.; et al. Identification and fine mapping of spot blotch (Bipolaris sorokiniana) resistance gene Sb4 in wheat. Theor. Appl. Genet. 2020, 133, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Maddox, C.E.; Laur, L.M.; Tian, L. Antibacterial activity of phenolic compounds against the phytopathogen Xylella fastidiosa. Curr. Microbiol. 2010, 60, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treutter, D. Signifificance of flflavonoids in plant resistance and enhancement of their biosynthesis. Plant Biol. 2005, 7, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Hammerbacher, A.; Raguschke, B.; Wright, L.P.; Gershenzon, J. Gallocatechin biosynthesis via a flflavonoid 3’,5’-hydroxylase is a defense response in Norway spruce against infection by the bark beetle-associated sapstaining fungus Endoconidiophora polonica. Phytochemistry 2018, 148, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Cushnie, T.P.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Niculaes, C.; Abramov, A.; Hannemann, L.; Frey, M. Plant protection by benzoxazinoids—Recent insights into biosynthesis and function. Agronomy 2018, 8, 143. [Google Scholar] [CrossRef] [Green Version]
- Fabro, G.; Kovács, I.; Pavet, V.; Szabados, L.; Alvarez, M.E. Proline accumulation and AtP5CS2 gene activation are induced by plant-pathogen incompatible interactions in Arabidopsis. Mol. Plant Microbe Interact. 2004, 17, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Cecchini, N.M.; Monteoliva, M.I.; Alvarez, M.E. Proline dehydrogenase contributes to pathogen defense in Arabidopsis. Plant Physiol. 2011, 155, 1947–1959. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.; Funck, D.; Szabados, L.; Rentsch, D. Proline metabolism and transport in plant development. Amino Acids 2010, 39, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Szabados, L.; Savouré, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef]
- Ren, Z.; Muhae-Ud-Din, G.; Liu, J.; Liu, T.; Chen, W.; Gao, L. Metabolomics Analysis of Tilletia Controversa Kühn Infected and Non-infected Grains of Wheat. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Ranjan, A.; Westrick, N.M.; Jain, S.; Piotrowski, J.S.; Ranjan, M.; Kessens, R.; Stiegman, L.; Grau, C.R.; Conley, S.P.; Smith, D.L.; et al. Resistance against Sclerotinia sclerotiorum in soybean involves a reprogramming of the phenylpropanoid pathway and up-regulation of antifungal activity targeting ergosterol biosynthesis. Plant Biotechnol. J. 2019, 17, 1567–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ube, N.; Harada, D.; Katsuyama, Y.; Osaki-Oka, K.; Tonooka, T.; Ueno, K.; Taketa, S.; Ishihara, A. Identification of phenylamide phytoalexins and characterization of inducible phenylamide metabolism in wheat. Phytochemistry 2019, 167, 112098. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Shen, S.; Wang, D.; Zhang, F.; Zhang, C.; Wang, Z.; Zhou, Q.; Wang, R.; Tao, H.; He, F.; et al. A monocot-specific hydroxycinnamoylputrescine gene cluster contributes to immunity and cell death in rice. Sci. Bull. 2021, 66, 2381–2393. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species of Metabolites | Number of Metabolites |
|---|---|
| amino acids and their derivatives | 95 |
| phenolic acids | 178 |
| nucleotides and their derivatives | 67 |
| flavone | 234 |
| lignans and coumarins | 30 |
| blending quality | 3 |
| alkaloid | 111 |
| terpenoids | 22 |
| organic acid | 84 |
| lipid | 154 |
| other | 100 |
| total: | 1078 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Huang, T.; Wang, Q.; Guo, Y.; Zhang, P.; Xie, H.; Liu, J.; Li, L.; Zhang, C.; Qin, P. Mechanisms of Resistance to Spot Blotch in Yunnan Iron Shell Wheat Based on Metabolome and Transcriptomics. Int. J. Mol. Sci. 2022, 23, 5184. https://doi.org/10.3390/ijms23095184
Zhang X, Huang T, Wang Q, Guo Y, Zhang P, Xie H, Liu J, Li L, Zhang C, Qin P. Mechanisms of Resistance to Spot Blotch in Yunnan Iron Shell Wheat Based on Metabolome and Transcriptomics. International Journal of Molecular Sciences. 2022; 23(9):5184. https://doi.org/10.3390/ijms23095184
Chicago/Turabian StyleZhang, Xuesong, Tingzhi Huang, Qianchao Wang, Yirui Guo, Ping Zhang, Heng Xie, Junna Liu, Li Li, Chuanli Zhang, and Peng Qin. 2022. "Mechanisms of Resistance to Spot Blotch in Yunnan Iron Shell Wheat Based on Metabolome and Transcriptomics" International Journal of Molecular Sciences 23, no. 9: 5184. https://doi.org/10.3390/ijms23095184