
Friend or Foe: The Relativity of (Anti)oxidative Agents and Pathways




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
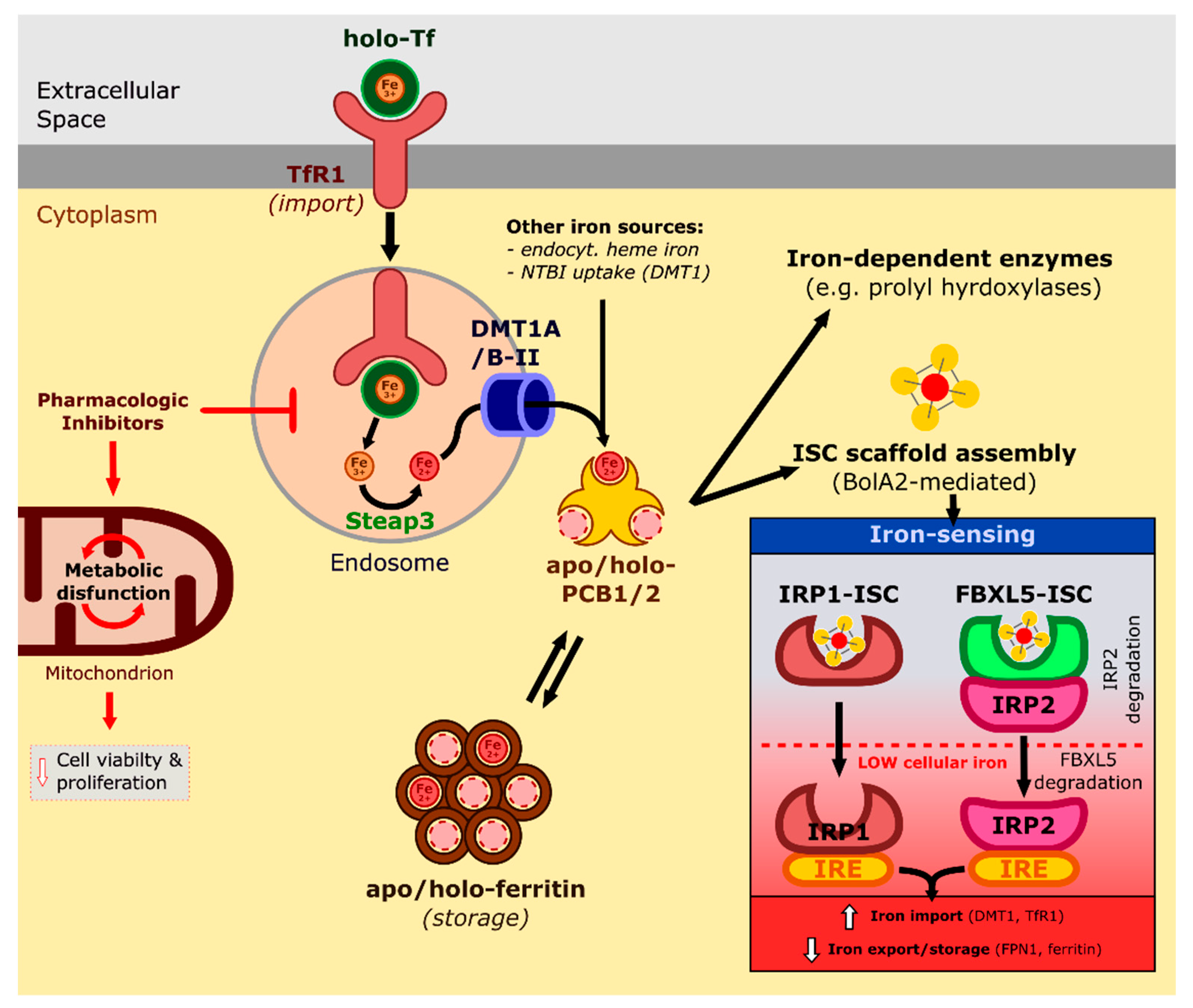
2. Iron, an Element with Multiple Roles
2.1. Absorption, Transport, and Cellular Uptake of Iron
2.2. Cellular Iron Homeostasis: Regulation and Dysregulation
2.3. Ferritinophagy and Iron Overload-Induced Toxicity
3. The Dual Role of ROS
3.1. Cellular Sources and Roles of ROS
3.2. ROS in Cancer
3.3. ROS in Ischemia-Reperfusion Injury
3.4. ROS in the Defence against Pathogens
4. Ascorbate: The Molecule of Clichés
4.1. The Excellent Antioxidant
4.2. How can Ascorbic Acid Give Solutions to Everyday Problems?
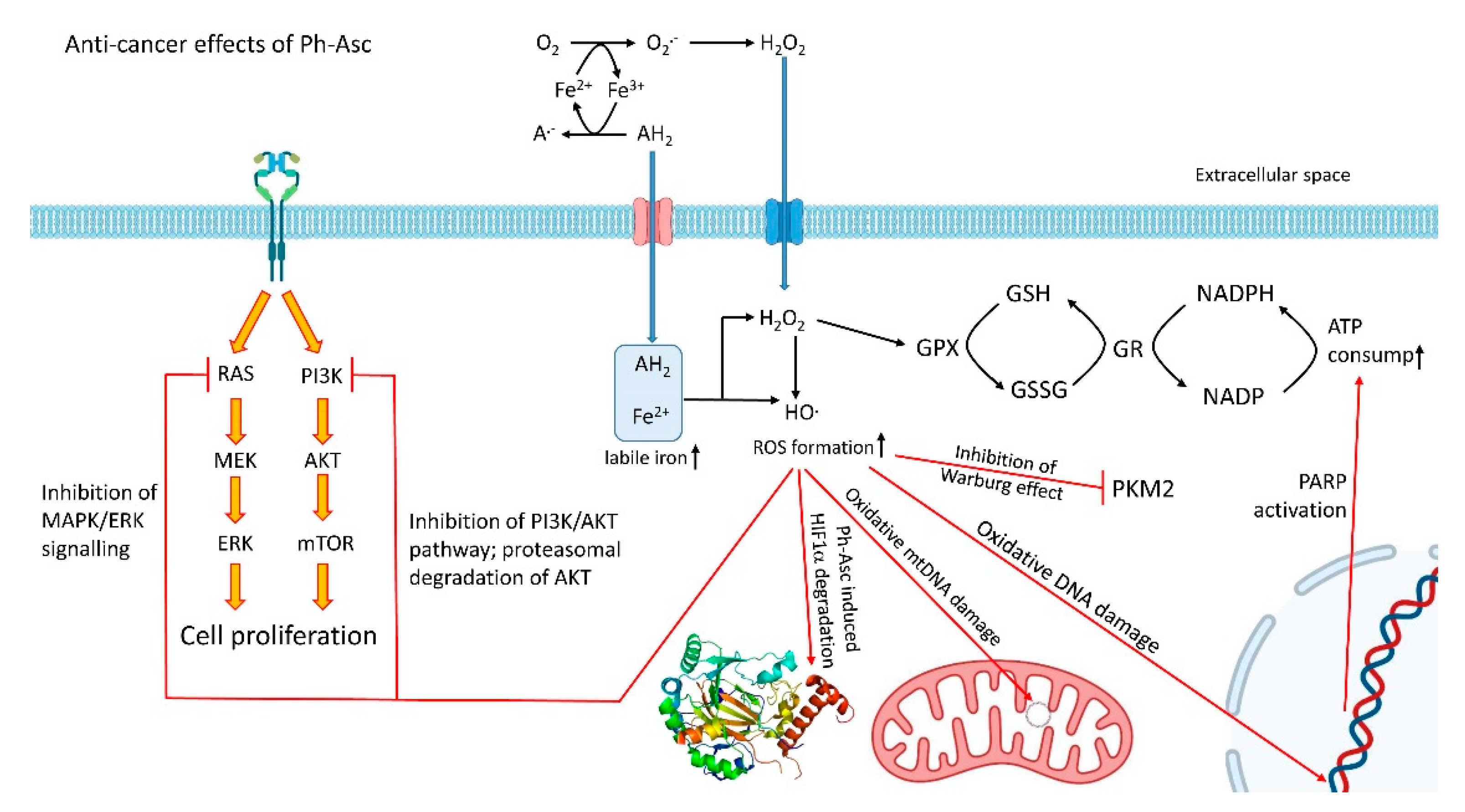
4.3. Outside of the Cliché: The Pro-Oxidant Role of Ascorbate
4.3.1. Rupture of Bioenergetics
4.3.2. DNA Damage
4.3.3. Accelerated Degradation of HIF-1α, Decreased Adaptation to Hypoxia
4.3.4. Impairment of Hybrid and Warburg Metabolism
4.3.5. The Halt of MAPK/ERK and PI3K/AKT Signalling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Knutson, M.D. Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice. Hepatology 2013, 58, 788–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanatori, I.; Kishi, F. DMT1 and iron transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (DCYTB) in Iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Liu, W.; Zhang, S.; Liu, S. The cardinal roles of ferroportin and its partners in controlling cellular iron in and out. Life Sci. 2020, 258, 118135. [Google Scholar] [CrossRef]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.V.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H.; et al. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol. Cell 2020, 77, 645–655.e7. [Google Scholar] [CrossRef]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.J.; Frey, A.G.; Palenchar, D.J.; Achar, S.; Bullough, K.Z.; Vashisht, A.; Wohlschlegel, J.A.; Philpott, C.C. A PCBP1–BolA2 chaperone complex delivers iron for cytosolic [2Fe–2S] cluster assembly. Nat. Chem. Biol. 2019, 15, 872–881. [Google Scholar] [CrossRef]
- Terzi, E.M.; Sviderskiy, V.O.; Alvarez, S.W.; Whiten, G.C.; Possemato, R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 2021, 7, eabg4302. [Google Scholar] [CrossRef]
- Xu, M.M.; Wang, J.; Xie, J.X. Regulation of iron metabolism by hypoxia-inducible factors. Sheng Li Xue Bao 2017, 69, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 508, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Vaites, L.P.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Ryu, M.S.; Zhang, D.; Protchenko, O.; Shakoury-Elizeh, M.; Philpott, C.C. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Investig. 2017, 127, 1786–1797. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Dolma, S.; Lessnick, S.L.; Hahn, W.C. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Ajoolabady, A.; Demillard, L.J.; Yu, W.; Hilaire, M.L.; Zhang, Y.; Ren, J. Dysregulation of iron metabolism in cardiovascular diseases: From iron deficiency to iron overload. Biochem. Pharmacol. 2021, 190, 114661. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; de Lima Barros, P.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, T.; Jemnitz, K.; Kardon, T.; Mandl, J.; Szarka, A. Ferroptosis is Involved in Acetaminophen Induced Cell Death. Pathol. Oncol. Res. 2015, 21, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-W.; Norwitz, S.G.; Norwitz, E.R. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyokuni, S.; Yanatori, I.; Kong, Y.; Zheng, H.; Motooka, Y.; Jiang, L. Ferroptosis at the crossroads of infection, aging and cancer. Cancer Sci. 2020, 111, 2665–2671. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cao, Y.; Cao, W.; Jia, Y.; Lu, N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020, 159, 104919. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raballand, V.; Benedikt, J.; Wunderlich, J.; von Keudell, A. Inactivation of Bacillus atrophaeus and of Aspergillus niger using beams of argon ions, of oxygen molecules and of oxygen atoms. J. Phys. D Appl. Phys. 2008, 41, 115207. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Gul, M.; Alzweiri, M.; Ishaqat, A.; ALSalamat, H.A.; Bashatwah, R.M. Reactive Oxygen Species: The Dual Role in Physiological and Pathological Conditions of the Human Body. Eurasian J. Med. 2018, 50, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Mumbengegwi, D.R.; Li, Q.; Li, C.; Bear, C.E.; Engelhardt, J.F. Evidence for a Superoxide Permeability Pathway in Endosomal Membranes. Mol. Cell. Biol. 2008, 28, 3700–3712. [Google Scholar] [CrossRef] [Green Version]
- Szarka, A.; Kapuy, O.; Lőrincz, T.; Bánhegyi, G. Vitamin C and Cell Death. Antioxid. Redox Signal. 2021, 34, 831–844. [Google Scholar] [CrossRef]
- Tóth, S.Z.; Lőrincz, T.; Szarka, A. Concentration Does Matter: The Beneficial and Potentially Harmful Effects of Ascorbate in Humans and Plants. Antioxid. Redox Signal. 2018, 29, 1516–1533. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Sokolovski, S.G.; Rafailov, E.U.; Abramov, A.Y.; Angelova, P.R. Singlet oxygen stimulates mitochondrial bioenergetics in brain cells. Free Radic. Biol. Med. 2021, 163, 306–313. [Google Scholar] [CrossRef]
- Onyango, A.N. Endogenous generation of singlet oxygen and ozone in human and animal tissues: Mechanisms, biological significance, and influence of dietary components. Oxid. Med. Cell. Longev. 2016, 2016, 2398573. [Google Scholar] [CrossRef] [Green Version]
- Dogra, V.; Kim, C. Singlet Oxygen Metabolism: From Genesis to Signaling. Front. Plant Sci. 2020, 10, 1640. [Google Scholar] [CrossRef] [Green Version]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z. Bin Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Sznarkowska, A.; Kostecka, A.; Meller, K.; Bielawski, K.P. Inhibition of cancer antioxidant defense by natural compounds. Oncotarget 2017, 8, 15996–16016. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.H.; Griffiths, H.R. The dual role of Reactive Oxygen Species in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic. Biol. Med. 2018, 125, 62–71. [Google Scholar] [CrossRef]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in favor of Toxic Effects. Oxid. Med. Cell. Longev. 2016, 2016, 8629024. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Czobor, Á.; Hajdinák, P.; Szarka, A. Rapid ascorbate response to bacterial elicitor treatment in Arabidopsis thaliana cells. Acta Physiol. Plant. 2017, 39, 62. [Google Scholar] [CrossRef]
- Czobor, Á.; Hajdinák, P.; Németh, B.; Piros, B.; Németh, Á.; Szarka, A. Comparison of the response of alternative oxidase and uncoupling proteins to bacterial elicitor induced oxidative burst. PLoS ONE 2019, 14, e0210592. [Google Scholar] [CrossRef] [PubMed]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hajdinák, P.; Czobor, Á.; Szarka, A. The potential role of acrolein in plant ferroptosis-like cell death. PLoS ONE 2019, 14, e0227278. [Google Scholar] [CrossRef] [Green Version]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.; Lee, S. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Sabet Sarvestani, F.; Azarpira, N.; Al-Abdullah, I.H.; Tamaddon, A.M. microRNAs in liver and kidney ischemia reperfusion injury: Insight to improve transplantation outcome. Biomed. Pharmacother. 2021, 133, 110944. [Google Scholar] [CrossRef]
- Dar, W.A.; Sullivan, E.; Bynon, J.S.; Eltzschig, H.; Ju, C. Ischaemia reperfusion injury in liver transplantation: Cellular and molecular mechanisms. Liver Int. 2019, 39, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.; Evelson, P.; Gelpi, R.J. Protecting the heart from ischemia/reperfusion injury. Curr. Opin. Cardiol. 2017, 32, 784–790. [Google Scholar] [CrossRef]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.-M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Jia, P.; Chen, N.; Fang, Y.; Liang, Y.; Guo, M.; Ding, X. Delayed Remote Ischemic Preconditioning ConfersRenoprotection against Septic Acute Kidney Injury via Exosomal miR-21. Theranostics 2019, 9, 405–423. [Google Scholar] [CrossRef]
- Graham, D.B.; Becker, C.E.; Doan, A.; Goel, G.; Villablanca, E.J.; Knights, D.; Mok, A.; Ng, A.C.Y.; Doench, J.G.; Root, D.E.; et al. Functional genomics identifies negative regulatory nodes controlling phagocyte oxidative burst. Nat. Commun. 2015, 6, 7838. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 618. [Google Scholar] [CrossRef]
- Slauch, J.M. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Love, D.T.; Barrett, T.J.; White, M.Y.; Cordwell, S.J.; Davies, M.J.; Hawkins, C.L. Cellular targets of the myeloperoxidase-derived oxidant hypothiocyanous acid (HOSCN) and its role in the inhibition of glycolysis in macrophages. Free Radic. Biol. Med. 2016, 94, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Z.; Li, X.; Dong, G.; Zhang, M.; Xu, Z.; Yang, J. Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance. Mediat. Inflamm. 2020, 2020, 9254087. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Olsson, L.M.; Urbonaviciute, V.; Yang, M.; Bäckdahl, L.; Holmdahl, R. Association of NOX2 subunits genetic variants with autoimmune diseases. Free Radic. Biol. Med. 2018, 125, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef] [PubMed]
- Martner, A.; Aydin, E.; Hellstrand, K. NOX2 in autoimmunity, tumor growth and metastasis. J. Pathol. 2019, 247, 151–154. [Google Scholar] [CrossRef]
- Shen, J.; Griffiths, P.T.; Campbell, S.J.; Utinger, B.; Kalberer, M.; Paulson, S.E. Ascorbate oxidation by iron, copper and reactive oxygen species: Review, model development, and derivation of key rate constants. Sci. Rep. 2021, 11, 7417. [Google Scholar] [CrossRef]
- Buettner, G.R.; Schafer, F.Q. Ascorbate as an antioxidant. In Vitamin C; Asard, H., May, J.M., Smirnoff, N., Eds.; Taylor & Francis: London, UK, 2003; pp. 173–188. [Google Scholar]
- Szarka, A.; Tomasskovics, B.; Bánhegyi, G. The Ascorbate-glutathione-α-tocopherol Triad in Abiotic Stress Response. Int. J. Mol. Sci. 2012, 13, 4458–4483. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.G.; Park, D.H.; Chae, Y.C. Role of Mitochondrial Stress Response in Cancer Progression. Cells 2022, 11, 771. [Google Scholar] [CrossRef]
- Gruss-Fischer, T.; Fabian, I. Protection by ascorbic acid from denaturation and release of cytochrome c, alteration of mitochondrial membrane potential and activation of multiple caspases induced by H(2)O(2), in human leukemia cells. Biochem. Pharmacol. 2002, 63, 1325–1335. [Google Scholar] [CrossRef]
- Perez-Cruz, I.; Carcamo, J.M.; Golde, D.W. Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood 2003, 102, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Dhar-Mascareño, M.; Cárcamo, J.M.; Golde, D.W. Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in human endothelial cells are inhibited by vitamin C. Free Radic. Biol. Med. 2005, 38, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Takeda, Y.; Sakahara, S.; Ishii, N.; Kobayashi, S.; Abe, H.; Asao, H.; Fujii, J. Heterozygous SOD1 deficiency in mice with an NZW background causes male infertility and an aberrant immune phenotype. Free Radic. Res. 2019, 53, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Ryan, M.J.; Didion, L.A.; Fegan, P.E.; Sigmund, C.D.; Faraci, F.M. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ. Res. 2002, 91, 938–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J.; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, S.; Shimizu, T.; Shirasawa, T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J. Biol. Chem. 2006, 281, 31713–31719. [Google Scholar] [CrossRef]
- Iuchi, Y.; Okada, F.; Onuma, K.; Onoda, T.; Asao, H.; Kobayashi, M.; Fujii, J. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochem. J. 2007, 402, 219–227. [Google Scholar] [CrossRef]
- Busuttil, R.A.; Garcia, A.M.; Cabrera, C.; Rodriguez, A.; Suh, Y.; Kim, W.H.; Huang, T.T.; Vijg, J. Organ-specific increase in mutation accumulation and apoptosis rate in CuZn-superoxide dismutase-deficient mice. Cancer Res. 2005, 65, 11271–11275. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, S.; Kibe, N.; Kurahashi, T.; Fujii, J. Differential responses of SOD1-deficient mouse embryonic fibroblasts to oxygen concentrations. Arch. Biochem. Biophys. 2013, 537, 5–11. [Google Scholar] [CrossRef]
- Inoue, E.; Tano, K.; Yoshii, H.; Nakamura, J.; Tada, S.; Watanabe, M.; Seki, M.; Enomoto, T. SOD1 Is Essential for the Viability of DT40 Cells and Nuclear SOD1 Functions as a Guardian of Genomic DNA. J. Nucleic Acids 2010, 2010, 795946. [Google Scholar] [CrossRef] [Green Version]
- Tamari, Y.; Nawata, H.; Inoue, E.; Yoshimura, A.; Yoshii, H.; Kashino, G.; Seki, M.; Enomoto, T.; Watanabe, M.; Tano, K. Protective roles of ascorbic acid in oxidative stress induced by depletion of superoxide dismutase in vertebrate cells. Free Radic. Res. 2013, 47, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, T.; Takeda, Y.; Nakano, T.; Akatsuka, S.; Kinoshita, D.; Kurahashi, T.; Saitoh, S.; Yamada, K.; Miyata, S.; Asao, H.; et al. Defective biosynthesis of ascorbic acid in Sod1-deficient mice results in lethal damage to lung tissue. Free Radic. Biol. Med. 2021, 162, 255–265. [Google Scholar] [CrossRef]
- Sentman, M.L.; Granström, M.; Jakobson, H.; Reaume, A.; Basu, S.; Marklund, S.L. Phenotypes of mice lacking extracellular superoxide dismutase and copper- and zinc-containing superoxide dismutase. J. Biol. Chem. 2006, 281, 6904–6909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra-Flores, P.; Riquelme, J.A.; Valenzuela-Bustamante, P.; Leiva-Navarrete, S.; Vivar, R.; Cayupi-Vivanco, J.; Castro, E.; Espinoza-Pérez, C.; Ruz-Cortés, F.; Pedrozo, Z.; et al. The Association of Ascorbic Acid, Deferoxamine and N-Acetylcysteine Improves Cardiac Fibroblast Viability and Cellular Function Associated with Tissue Repair Damaged by Simulated Ischemia/Reperfusion. Antioxidants 2019, 8, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabani, P.; Ghazizadeh, Z.; Gorgani-Firuzjaee, S.; Molazem, M.; Rajabi, S.; Vahdat, S.; Azizi, Y.; Doosti, M.; Aghdami, N.; Baharvand, H. Cardioprotective effects of omega-3 fatty acids and ascorbic acid improve regenerative capacity of embryonic stem cell-derived cardiac lineage cells. Biofactors 2019, 45, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, R.C.; Archana, V.; Binu, P.; Arathi, P.; Nair, R.H. L-Ascorbic Acid and α-Tocopherol Reduces Hepatotoxicity Associated with Arsenic Trioxide Chemotherapy by Modulating Nrf2 and Bcl2 Transcription Factors in Chang liver Cells. Nutr. Cancer 2018, 70, 684–696. [Google Scholar] [CrossRef]
- Ludke, A.; Akolkar, G.; Ayyappan, P.; Sharma, A.K.; Singal, P.K. Time course of changes in oxidative stress and stress-induced proteins in cardiomyocytes exposed to doxorubicin and prevention by vitamin C. PLoS ONE 2017, 12, e0179452. [Google Scholar] [CrossRef] [Green Version]
- Parascandolo, A.; Laukkanen, M.O. Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1-3 Signal Transduction Pathways. Antioxid. Redox Signal. 2019, 30, 443–486. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L.; Leibovitz, B. Ascorbic acid and cancer: A review. Cancer Res. 1979, 39, 663–681. [Google Scholar]
- Buranasudja, V.; Doskey, C.M.; Gibson, A.R.; Wagner, B.A.; Du, J.; Gordon, D.J.; Koppenhafer, S.L.; Cullen, J.J.; Buettner, G.R. Pharmacologic Ascorbate Primes Pancreatic Cancer Cells for Death by Rewiring Cellular Energetics and Inducing DNA Damage. Mol. Cancer Res. 2019, 17, 2102–2114. [Google Scholar] [CrossRef]
- Cieslak, J.; Cullen, J. Treatment of Pancreatic Cancer with Pharmacological Ascorbate. Curr. Pharm. Biotechnol. 2015, 16, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra18. [Google Scholar] [CrossRef] [PubMed]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H 2 O 2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sendoel, A.; Kohler, I.; Fellmann, C.; Lowe, S.W.; Hengartner, M.O. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010, 465, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.A.; Vasile, E.; Feng, D.; Sundberg, C.; Brown, L.F.; Detmar, M.J.; Lawitts, J.A.; Benjamin, L.; Tan, X.; Manseau, E.J.; et al. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 2002, 196, 1497–1506. [Google Scholar] [CrossRef]
- Brown, L.F.; Detmar, M.; Claffey, K.; Nagy, J.A.; Feng, D.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A multifunctional angiogenic cytokine. In Regulation of Angiogenesis; Experientia Supplementum EXS; Springer: Berlin, Germany, 1997; Volume 79, pp. 233–269. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Klein, E.; Drebber, U.; Baldus, S.E.; Cooc, J.; Azuma, M.; Metzger, R.; et al. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 2008, 10, 674–679. [Google Scholar] [CrossRef] [Green Version]
- Mandl, J.; Szarka, A.; Bánhegyi, G. Vitamin C: Update on physiology and pharmacology. Br. J. Pharmacol. 2009, 157, 1097–1110. [Google Scholar] [CrossRef] [Green Version]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature 2012, 487, 380–384. [Google Scholar] [CrossRef]
- Wilkes, J.G.; O’Leary, B.R.; Du, J.; Klinger, A.R.; Sibenaller, Z.A.; Doskey, C.M.; Gibson-Corley, K.N.; Alexander, M.S.; Tsai, S.; Buettner, G.R.; et al. Pharmacologic ascorbate (P-AscH−) suppresses hypoxia-inducible Factor-1α (HIF-1α) in pancreatic adenocarcinoma. Clin. Exp. Metastasis 2018, 35, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Kapuy, O.; Makk-merczel, K.; Szarka, A. Therapeutic Approach of KRAS Mutant Tumours by the Combination of Pharmacologic Ascorbate and Chloroquine. Biomolecules 2021, 11, 652. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, J.; Ren, D.; Tsai, W.L.; Li, F.; Amanullah, A.; Hudson, T. Probing of C-terminal lysine variation in a recombinant monoclonal antibody production using Chinese hamster ovary cells with chemically defined media. Biotechnol. Bioeng. 2012, 109, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Unterlass, J.E.; Curtin, N.J. Warburg and Krebs and related effects in cancer. Expert Rev. Mol. Med. 2019, 21, e4. [Google Scholar] [CrossRef] [PubMed]
- Serna-Blasco, R.; Sanz-Álvarez, M.; Aguilera, Ó.; García-Foncillas, J. Targeting the RAS-dependent chemoresistance: The Warburg connection. Semin. Cancer Biol. 2019, 54, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Shen, Z.; Yang, Q.; Sui, F.; Pu, J.; Ma, J.; Ma, S.; Yao, D.; Ji, M.; Hou, P. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 2019, 9, 4461–4473. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Li, P.; Han, B.; Jia, H.; Liang, Q.; Wang, H.; Gu, M.; Cai, J.; Li, S.; Zhou, Y.; et al. Vitamin C sensitizes BRAFV600E thyroid cancer to PLX4032 via inhibiting the feedback activation of MAPK/ERK signal by PLX4032. J. Exp. Clin. Cancer Res. 2021, 40, 34. [Google Scholar] [CrossRef]
- Balihodzic, A.; Prinz, F.; Dengler, M.A.; Calin, G.A.; Jost, P.J.; Pichler, M. Non-coding RNAs and ferroptosis: Potential implications for cancer therapy. Cell Death Differ. 2022. ahead of print. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szarka, A.; Lőrincz, T.; Hajdinák, P. Friend or Foe: The Relativity of (Anti)oxidative Agents and Pathways. Int. J. Mol. Sci. 2022, 23, 5188. https://doi.org/10.3390/ijms23095188
Szarka A, Lőrincz T, Hajdinák P. Friend or Foe: The Relativity of (Anti)oxidative Agents and Pathways. International Journal of Molecular Sciences. 2022; 23(9):5188. https://doi.org/10.3390/ijms23095188
Chicago/Turabian StyleSzarka, András, Tamás Lőrincz, and Péter Hajdinák. 2022. "Friend or Foe: The Relativity of (Anti)oxidative Agents and Pathways" International Journal of Molecular Sciences 23, no. 9: 5188. https://doi.org/10.3390/ijms23095188
APA StyleSzarka, A., Lőrincz, T., & Hajdinák, P. (2022). Friend or Foe: The Relativity of (Anti)oxidative Agents and Pathways. International Journal of Molecular Sciences, 23(9), 5188. https://doi.org/10.3390/ijms23095188