Abstract
Nonalcoholic steatohepatitis (NASH) is a chronic liver disease that increases cardiovascular disease risk. Indoleamine 2,3-dioxygenase-1 (IDO1)-mediated tryptophan (Trp) metabolism has been proposed to play an immunomodulatory role in several diseases. The potential of IDO1 to be a link between NASH and cardiovascular disease has never been investigated. Using Apoe−/− and Apoe−/−Ido1−/− mice that were fed a high-fat, high-cholesterol diet (HFCD) to simultaneously induce NASH and atherosclerosis, we found that Ido1 deficiency significantly accelerated atherosclerosis after 7 weeks. Surprisingly, Apoe−/−Ido1−/− mice did not present a more aggressive NASH phenotype, including hepatic lipid deposition, release of liver enzymes, and histopathological parameters. As expected, a lower L-kynurenine/Trp (Kyn/Trp) ratio was found in the plasma and arteries of Apoe−/−Ido1−/− mice compared to controls. However, no difference in the hepatic Kyn/Trp ratio was found between the groups. Hepatic transcript analyses revealed that HFCD induced a temporal increase in tryptophan 2,3-dioxygenase (Tdo2) mRNA, indicating an alternative manner to maintain Trp degradation during NASH development in both Apoe−/− and Apoe−/−Ido1−/mice−. Using HepG2 hepatoma cell and THP1 macrophage cultures, we found that iron, TDO2, and Trp degradation may act as important mediators of cross-communication between hepatocytes and macrophages regulating liver inflammation. In conclusion, we show that Ido1 deficiency aggravates atherosclerosis, but not liver disease, in a newly established NASH and atherosclerosis comorbidity model. Our data indicate that the overexpression of TDO2 is an important mechanism that helps in balancing the kynurenine pathway and inflammation in the liver, but not in the artery wall, which likely determined disease outcome in these two target tissues.
1. Introduction
Atherosclerosis is the underlying cause of most cardiovascular diseases (CVDs) and the leading cause of morbidity and mortality worldwide [1,2]. Atherosclerosis development is influenced by several risk factors, including dyslipidaemia, hypertension, smoking, and diabetes; targeting these risk factors is currently the main strategy to combat the CVD burden. Despite all the recent developments in medicine, only up to one-third of major clinical consequences of atherosclerosis, e.g., myocardial infarction, seem to be prevented by using the current guidelines [3].
It is now well recognized that atherosclerosis is a chronic inflammatory disease that is likely initiated by the accumulation of low-density lipoprotein (LDL) in the artery wall. The accumulation and modifications of LDL trigger maladaptive innate and adaptive immune responses in the artery wall, driving the formation of an atherosclerotic plaque, as well as a fibrous cap that, upon rupture, can cause thrombosis and a CVD event [4]. Large preclinical and, more recently, clinical evidence indicate that immunomodulation of vascular inflammation could be used to reduce the CVD burden beyond current guidelines for handling classic disease risk factors [5].
Activation of the immune system is not only well established within the pathophysiology of atherosclerosis, but also plays a major role in the development of nonalcoholic fatty liver disease (NAFLD), which can progress to nonalcoholic steatohepatitis (NASH); the latter that is defined as the combination of liver steatosis, parenchymal damage (hepatocyte apoptosis and ballooning, and focal necrosis), lobular and/or portal inflammation, and variable degrees of fibrosis [6,7]. Due to its association with obesity, type 2 diabetes, and the ectopic accumulation of lipids in the liver parenchyma, NASH has been considered an advanced hepatic component of metabolic syndrome and an additional risk factor for CVD [7].
Indoleamine 2,3-dioxygenase-1 (IDO1), the rate-limiting enzyme of the kynurenine pathway of tryptophan (Trp) metabolism, has been identified as a key immunomodulatory enzyme implicated in different diseases, including atherosclerosis and liver disease. It has been proposed that the local depletion of Trp and/or the production of potent bioactive metabolites of this pathway, collectively known as kynurenines, can modulate immune cell functions such as activation, proliferation, and migration [8].
While increased IDO1 expression, in the great majority of cases, has been implicated in atheroprotection [8,9,10,11,12,13,14,15,16], its role in liver inflammation and NAFLD/NASH is less clear. It has been shown that, upon high-fat diet feeding or the injection of CCL4, Ido1-deficient mice present increased hepatic inflammation and fibrosis [17,18]. IDO-dependent regulation of IL-17 release has been suggested as a major mechanism attenuating liver fibrosis [19,20]. However, IDO expression has also been associated with obesity and worsening insulin resistance through the regulation of intestinal permeability, which also influences liver steatosis [21].
Although NAFLD/NASH-related mortality is usually linked to adverse hepatic outcomes such as cirrhosis, liver failure, and hepatocellular carcinoma, CVDs are the main cause of mortality among patients with liver disease [22,23]. Thus, it has been hypothesized that NAFLD/NASH-related steatosis is a chronic inducer of low-grade hepatic inflammation and the source of several immunomodulatory mediators, which, when systemically released, could accelerate vascular inflammation and the development of CVDs [24]. Yet, the precise molecular mechanisms underlying the relationship between NASH and atherosclerosis remain unclear.
In this study, we validated a new murine model that develops both NASH and atherosclerosis. Using Apoe−/− and Apoe−/−Ido1−/− mice that were fed a high-fat and cholesterol-rich diet, we demonstrate an essential role of IDO1 in accelerating vascular but not liver disease in the same animals. Our results indicate that disease-related factors promote the upregulation of tryptophan 2,3-dioxygenase (Tdo2) in the liver, which could help maintain local Trp degradation and prevent the aggravation of NASH.
2. Results
2.1. Apoe−/− Is a Suitable Strain for Studying Atherosclerosis and NASH as Comorbidities—Model Validation
Both male and female Apoe−/− mice were fed HFCD for 3.5 and 7.0 weeks, and systemic organ-specific changes were evaluated. As expected, HFCD feeding led to significantly accelerated atherosclerosis, and after 7.0 weeks, a twofold increase in plaque area in the aortic arch was observed compared to chow-fed mice (Table 1; Figure 1A,B). A systematic review of the literature showed that, in the context of experimental atherosclerosis, a higher plaque burden is seen in young female hyperlipidaemic mice compared with their male counterparts [25]. Although we observed a trend towards females developing larger lesions than males after 7.0 weeks of HFCD, no significant difference was observed. No significant difference in lesion size between the HFCD- and chow-fed groups was observed at the 3.5-week time point (Table 1).

Table 1.
Characterization of HFCD-induced vascular and liver disease as comorbidities.
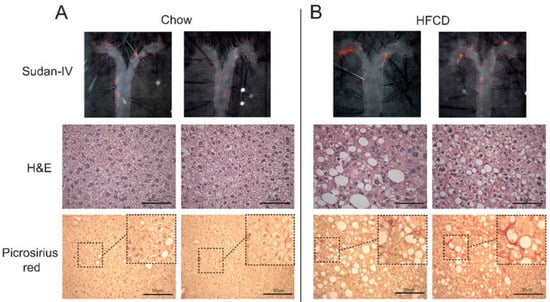
Figure 1.
Characterization of the NASH and atherosclerosis dual model. Apoe−/− mice were fed chow or a high-fat-cholesterol diet (HFCD) for 7 weeks, and representative pictures of atherosclerotic disease and NASH are shown; complete descriptive data of chow- and HFCD-fed mice are shown in Table 1. Top panels show en face lipid staining with Sudan-IV; middle panels show H&E staining showing hepatocyte ballooning; and the bottom panels show collagen deposition detected by picrosirius red staining in (A) chow- and (B) HFCD-fed Apoe−/− mice. Bar = 50 µm.
In line with the atherosclerosis data, HFCD-fed mice presented significantly higher levels of plasma cholesterol and triglycerides, especially after 7.0 weeks of diet feeding (Table 1). Although not significant within 7 weeks, HFCD presented a clear trend towards a faster weight gain than chow-fed mice (Supplementary Figure S1).
After confirming the atherosclerotic phenotype in our mice, we evaluated the effects of HFCD feeding on liver-related parameters. The liver-to-body ratio was significantly increased in Apoe−/− mice after 3.5 and 7.0 weeks of HFCD feeding (Table 1). These results were paralleled by significantly increased levels of plasma AST and ALT (Table 1). Additionally, hepatic accumulation of free iron was significantly increased in HFCD-fed mice compared to controls at the 7.0 weeks’ time point (Table 1).
Altogether, the previous results indicated that HFCD feeding of Apoe−/− mice promoted liver damage. Corroborating the latter affirmation, 7.0 weeks of HFCD feeding increased the hepatic levels of TNF and CCL2 (Table 1). Interestingly, the hepatic levels of IL-10 followed a different pattern, and substantially lower levels of the cytokine were seen after 7.0 weeks, which was independent of the diet (Table 1).
Considering that atherosclerosis and the first signs of NASH were observed after 7.0 weeks of HFCD, we selected this time point for further analyses. At this time point, HFCD clearly promoted hepatocyte ballooning (Figure 1A,B) and increased the accumulation of collagen, as evidenced by picrosirius red staining (Figure 1A,B). Further analyses showed that HFCD-fed mice presented significantly higher levels of hepatic cholesterol and triglycerides (Table 1), which was followed by a close to significant increase in hepatic hydroxyproline (Table 1) and a significant increase in Col1a1 mRNA levels, which encodes for the pro-alpha1 chain of type I collagen (Table 1).
2.2. Genetic Ablation of IDO1 in Apoe−/− Mice Accelerates Vascular, but Not Liver, Disease
Our data indicated that 7.0 weeks of HFCD feeding represents a suitable time point to study both atherosclerosis and NASH as concomitant diseases, and this protocol was selected to evaluate the role of IDO1 in disease. In line with our previous studies [9,10], ablation of IDO activity (Apoe−/−Ido1−/−) significantly increased atherosclerosis in the aortic arch compared to Apoe−/− mice (Figure 2A,B). Hence, Apoe−/−Ido1−/− mice also presented more lesions in the aortic root and increased Mac-2+ macrophage infiltration compared to Apoe−/− controls (Figure 2C,D).
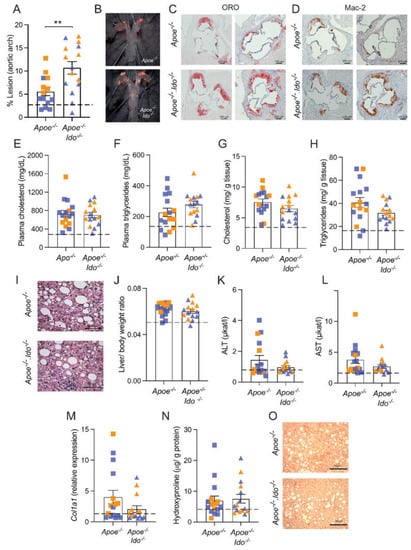
Figure 2.
Effects of IDO1 genetic ablation on NASH and atherosclerosis development. Apoe−/− or Apoe−/−Ido1−/− mice were fed a high-fat cholesterol diet (HFCD) for 7 weeks; pooled data from two independent experiments are shown. (A) Quantification of en face Sudan IV-stained aortic arches (n = 14–15). (B) Representative pictures of the aortic arches. (C) Representative pictures of the atherosclerotic burden stained by ORO in aortic root sections of Apoe−/− or Apoe−/− Ido1−/− mice (n = 2/group). (D) Representative picture of Mac-2+ macrophage infiltration in the aortic roots of Apoe−/− or Apoe−/− Ido1−/− mice (n = 2/group). Panel (E) shows total cholesterol and (F) triglyceride levels in plasma (n = 15–16). (G,H) Total levels of cholesterol and triglycerides in Apoe−/− and Apoe−/−Ido1−/− mice livers (n = 15–16). (I) Representative pictures of H&E-stained liver sections. (J) Liver/body ratio (n = 15–16), (K,L) plasma ALT and AST levels (n = 15–16). (M) Relative hepatic collagen (Col1a1) mRNA expression (n = 15–16) and (N) hepatic hydroxyproline levels (n = 15–16). (O) Representative pictures of picrosirius red-stained liver sections. ** p < 0.01; differences were detected using the Mann–Whitney U test; dotted lines refer to baseline levels of Apoe−/− mice fed a chow diet for 7 weeks (Table 1); orange and blue colours are used to identify female and male mice, respectively. Bar = 50 µm.
Next, we evaluated whether IDO1 ablation would impact NASH-related parameters in our model. There was no difference in plasma cholesterol and triglycerides (Figure 2E,F) or bodyweight between groups (Supplementary Figure S2). In line with the plasma data, Apoe−/− and Apoe−/−Ido1−/− mice presented no difference in the hepatic accumulation of cholesterol and triglyceride levels (Figure 2G,H), and similar hepatocyte ballooning was observed between groups (Figure 2I). Further analyses showed that Apoe−/− and Apoe−/−Ido1−/− also did not differ in terms of the liver-to-body weight ratio (Figure 2J), plasma levels of ALT and AST (Figure 2K,L), hepatic Col1a1 mRNA levels (Figure 2M), and hydroxyproline content (Figure 2N). Corroborating with the latter result, no difference in the picrosirius red staining of collagen was observed between groups (Figure 2O).
IDO1 ablation is usually followed by increased inflammation in different disease models [26,27], including atherosclerosis [8,9,10]. Immunofluorescence and macrophage-related transcript analyses revealed that Apoe−/− and Apoe−/−Ido−/− mice presented no differences in the hepatic infiltration of macrophages and the mRNA levels for the Kupfer cell marker Clec4f (Figure 3A–D) and no clear shift towards M1- or M2-like macrophage polarization patterns (Figure 3E,F). Of note, M1 and M2 terminologies are an oversimplification of a vast repertoire of phenotypes that can develop within an inflamed tissue, including atherosclerosis [28].
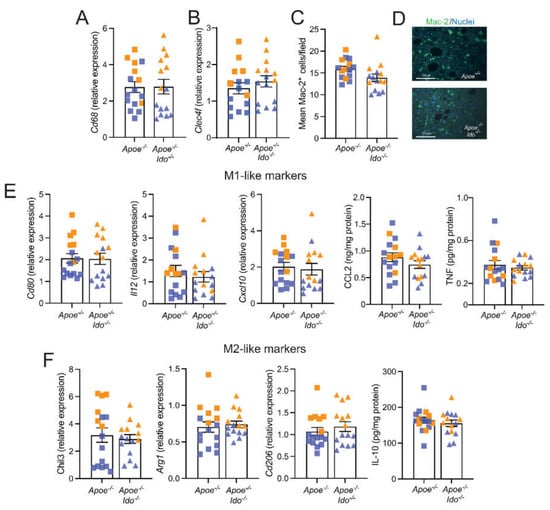
Figure 3.
Effects of IDO1 genetic ablation on liver inflammation. Apoe−/− or Apoe−/−Ido1−/− mice were fed a high-fat cholesterol diet (HFCD) for 7 weeks; pooled data from two independent experiments are shown. (A) Relative Cd68 mRNA expression (n = 15–16); (B) Relative Clec4f mRNA expression (n = 15); (C) Quantification of immunofluorescently stained Mac-2+ macrophages (n = 15); and (D) representative Mac-2+ fluorescence staining of liver from Apoe−/− or Apoe−/−Ido1−/−; Bar = 50 µm. (E) Relative hepatic mRNA expression of M1-like macrophage markers (Cd80, Il12, and Cxcl10 mRNA, and CCL2 and TNF protein) (n = 15–16) and (F) M2-like macrophage markers (Chil3, Arg1, and Cd206 mRNA, and IL-10 protein) (n = 15–16). Orange and blue colours are used to identify female and male mice, respectively; no differences between groups were detected using a Mann–Whitney U test.
2.3. Apoe−/−Ido1−/− Mice Presented Intact Hepatic Trp Degradation Rates despite Reduced Systemic and Aortic Trp Degradation Rates
The Kyn/Trp is used as a surrogate marker of IDO1 activity and the degradation of Trp within the kynurenine pathway. As expected, the Kyn/Trp ratio was reduced in plasma and aortas from Apoe−/−Ido1−/− mice compared to Apoe−/− mice (Figure 4A,B). Unexpectedly, no difference in the hepatic Kyn/Trp ratio was observed between the groups (Figure 4C). In line with the fact that increased rates of Trp degradation are usually associated with decreased inflammation, we found that the aortic Kyn/Trp ratio was inversely correlated with the percentage of lesions in the aortic arch (Figure 4D). Despite no difference between groups on Mac-2+ macrophage numbers, the hepatic Kyn/Trp ratio was also inversely correlated with the macrophage marker CD68 (Figure 4E), suggesting that hepatic inflammation could be regulated by the degree of Trp degradation in the liver.
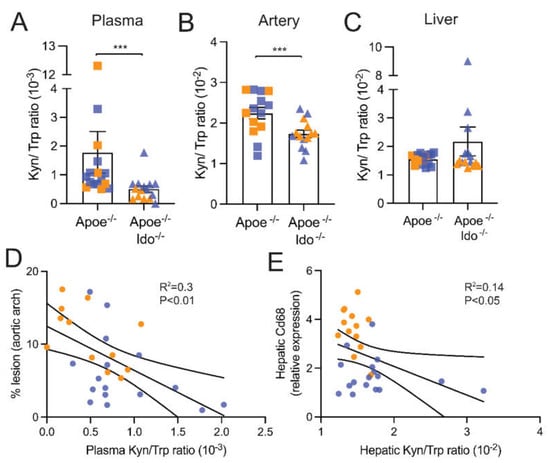
Figure 4.
Systemic and local tryptophan degradation rates. Apoe−/− or Apoe−/− Ido1−/− mice were fed a high-fat cholesterol diet (HFCD) for 7 weeks; pooled data from two independent experiments are shown. The L-kynrenine to Trp ratio (Kyn/Trp) in (A) plasma (n = 15–16), (B) artery homogenate (n = 14), and (C) liver homogenate (n = 15–16), was estimated using specific ELISA kits, as described in the methods. (D) shows the correlation between % lesion and Kyn/Trp ratio in the aorta. (E) shows the correlation between relative Cd68 mRNA and the Kyn/Trp ratio in the liver. *** p < 0.001; Differences were detected using the Mann–Whitney U test. Correlations were determined using simple linear regression.
2.4. HFCD Increases Hepatic TDO2 Expression
Although IDO1 has been implicated in the regulation of inflammation, another enzyme, tryptophan-2,3-dioxygenase (TDO2), is also involved in the first and rate-limiting step of the kynurenine pathway [29]. We observed a clear trend towards a temporal increase in the hepatic levels of Tdo2 mRNA between 3.5 and 7.0 weeks of HFCD feeding in Apoe−/−Ido1−/− mice (Figure 5). Interestingly, a similar increase in Tdo2 mRNA was observed in Apoe−/− (Figure 5). Although hepatic expression of TDO2 could be one explanation for the maintenance of Trp degradation and protection against the aggravation of liver disease in Apoe−/−Ido1−/−, the similar increase seen in the liver of Apoe−/− mice suggests that other mechanisms could influence Trp degradation in the presence of IDO1. Interestingly, aortic TDO2 protein levels were decreased in Apoe−/−Ido1−/− compared to Apoe-/- mice after 3.5 weeks of HFCD feeding, while no difference was observed between groups at the 7.0 weeks’ time point (Supplementary Figure S3).
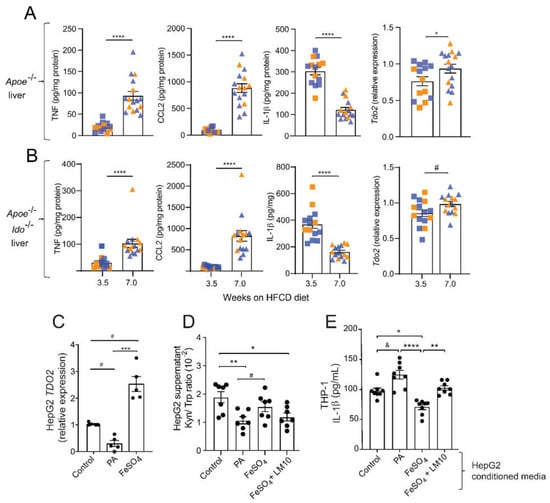
Figure 5.
Fatty acids and iron regulate TDO2-dependent Trp degradation and consequences for THP-1 macrophage activation. (A,B) TNF, CCL2, and IL-1β protein levels and relative Tdo2 mRNA expression in livers from Apoe−/− and Apoe−/−Ido1−/− mice fed a high-fat cholesterol diet (HFCD) for 3.5 and 7 weeks. (A,B) Pooled data from two independent experiments are shown; n = 14–15/group. © Relative expression of TDO2 mRNA in HepG2 cells treated with palmitic acid (PA, 500 μM) or iron (FeSO4, 100 μM) (n = 5). (D) Kyn/ Trp ratio in the supernatants of HepG2 cells treated with PA (500 μM), FeSO4, (100 μM), or FeSO4 + TDO2-inhibitor LM10 (0.62 μM) (n = 7). (E) IL-1β release from THP-1 macrophages pre-treated with conditioned media from HepG2 cells incubated with PA (500 μM), FeSO4 (100 μM), or FeSO4 + TDO2-inhibitor LM10 (0.62 μM); in addition, cells were stimulated with LPS (10 ng/mL, 4 h) and ATP (5 mM, 30 min) for activation of the inflammasome and IL-1β release (n = 8). & p < 0.06; # p < 0.08; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. (A) Differences were detected using the Mann–Whitney U test. (B,C) Differences were detected using a one-way ANOVA and Dunn’s post hoc test.
We have shown that the kynurenine pathway metabolism can regulate inflammasome activation and IL-1β secretion by macrophages [30]. In line with these data, IL-1β levels have been found to be increased in the plasma of Tdo2−/− mice injected with LPS [31]. In our comorbidity model, Apoe−/− and Apoe−/−Ido1−/− mice showed a time-dependent increase in the hepatic levels of TNF and CCL2, while a concomitant decrease in the hepatic levels of IL-1β was observed (Figure 5), suggesting that the latter could be regulated by TDO2.
An increased accumulation of lipids and free iron are well-known characteristics of NASH progression [32], which was also observed in our model (Table 1 and Figure 1). Interestingly, TDO2 is a tetrameric haemoprotein that requires Fe2+ for its full activation as other catalytic haemoproteins, and iron has been proposed to upregulate TDO2 mRNA expression levels [33]. Considering all the previous, we tested whether excess palmitic acid (PA) or iron (FeSO4) could regulate the expression of TDO2 in the liver hepatoma cell line HepG2, and whether TDO2-mediated Trp metabolism on hepatic cells could influence IL-1β secretion by macrophages.
Forty-eight hours of incubation of HepG2 cells with PA downregulated, while FeSO4 substantially increased, TDO2 mRNA levels (Figure 5C). Analyses of the supernatant of these cultures showed a decrease in the Kyn/Trp ratio in the supernatants of HepG2 cells treated with PA, while no changes were observed in cells treated with FeSO4 (Figure 5D). Interestingly, the concomitant addition of the TDO2 inhibitor LM10 to HepG2 cells treated with FeSO4 showed a reduced Kyn/Trp ratio compared to the control (Figure 5D).
Next, we tested whether the regulation of TDO2 expression on HepG2 cells, and reflected alterations in Kyn/Trp ratio, could, in a paracrine manner, influence the response of THP1-differentiated macrophages to secrete IL-1β in vitro. We observed that conditioned media from HepG2 cells cultured with PA, which reduced their TDO2 expression and Kyn/Trp ratio, increased the secretion of IL-1β by THP1 macrophages (Figure 5E). Contrary to the effects of PA, the conditioned media from FeSO4-treated HepG2 cells, which upregulated TDO2 and maintained an unchanged the Kyn/Trp ratio, significantly inhibited IL-1β secretion; these protective properties were lost when HepG2 cells concomitantly received FeSO4 and the TDO2 inhibitor LM10 (Figure 5E).
3. Discussion
NAFLD/NASH typically exists within the ‘‘milieu” of major diseases that play a central role in increasing the risk of CVD, including obesity, diabetes, and dyslipidaemia. Not surprisingly, myocardial infarction and stroke are highly prevalent in patients with metabolic liver disease [23]. Increasing our knowledge of the underlying mechanisms by which NAFLD/NASH accelerates atherosclerosis and increases cardiovascular risk can help improve the diagnosis and management of CVDs. In this study, we show that HFCD feeding promotes NASH and atherosclerosis in parallel in Apoe−/− mice, establishing a new viable dual comorbidity model. By feeding Apoe−/−Ido1−/− mice with HFCD, we show that IDO1-dependent Trp metabolism plays a distinctive role in regulating vascular versus fatty liver disease.
There have been numerous attempts to generate animal models, especially murine models, that can recapitulate the aetiology, natural history, and/or progression that are inherent to atherosclerosis or NAFLD/NASH [34,35]. In this context, the two most common hypercholesterolaemic mouse strains used to study atherosclerosis, Apoe−/− and Ldlr−/−, have been evaluated regarding their susceptibility to developing NASH. Schierwagen et al., (2015) showed that 7 weeks of HFCD feeding led Apoe−/− mice to develop several features common to human NASH, including hepatic steatosis, inflammation, and a moderate degree of fibrosis [36]. Bieghs et al., (2012) showed that Ldlr−/− mice present increased sensitivity to hepatic inflammation, apoptosis, and fibrosis after 12 weeks of HFCD compared to the human APOE2 knock-in mouse (APOE2ki) and C57BL/6 strains [37]. Despite the potential within these models, atherosclerosis has not been investigated in these studies.
To date, only a few studies have attempted to explore disease-modifying targets that could concomitantly influence NASH and CVD. Recently, van den Hoek et al., (2020) have shown that Ldlr−/−. Leiden mice develop NASH with progressive liver fibrosis, as well as atherosclerosis, upon 28 weeks of special high caloric diet feeding [38]. In our study, we established that HFCD feeding of Apoe−/− mice could also be a suitable strain for studying NASH and atherosclerosis simultaneously with a swift 7-week protocol. Thus, in addition to CVD, our mice presented all clinical signs that are characteristic of NASH, including liver steatosis, cytoskeletal damage (hepatocellular ballooning and increased levels of liver enzymes), inflammation, and a moderate degree of fibrosis, which, although not required for disease diagnosis, may indicate the aggravation of the disease state [39,40].
Inflammation is the major regulator of IDO1-dependent Trp metabolism in different cells and organs [41]. Increased IDO1 activity has been considered an important immune metabolic feedback mechanism regulating innate and adaptive immune cell responses [8]. Whether operating directly or indirectly, increased Trp metabolism through the kynurenine pathway has been linked with CVD because of its role in regulating vasculature [42], insulin resistance [30,43,44,45], or skewing of the gut microbiota [21]. Taking all previous knowledge into account, IDO1 emerged as an interesting target to be investigated in our dual model.
As we have previously shown using pharmacological and genetic approaches [9,10], IDO1 ablation increases vascular inflammation and accelerates atherosclerosis, which could now be reproduced using a HFCD. Unexpectedly, in the current study, we did not observe an acceleration of liver disease. In light of the fact that using a downstream metabolite of IDO in the kynurenine pathway, 3-hydroxyanthranilic acid (3-HAA), could regulate cholesterol synthesis as well as plasma and hepatic cholesterol levels [30], our new data might appear counterintuitive. While further research will be needed to fully understand the potential causes of these differences, some hypothetical lines of reasoning could be drawn.
We previously showed that 3-HAA mediated strong lipid-lowering effects in Ldlr−/− mice [30,46]. Hence, it was shown that genetic ablation of IDO in Ldlr−/− led to a significant increase in plasma lipids [47]. When using Apoe−/− mice, the pharmacological inhibition of IDO promoted only mild alterations to their lipoprotein profile, while four weeks of treatment with 3-HAA did not reverse the effects of IDO1 inhibition on lipids [46]. Interestingly, the original work from Cole et al., (2015) showed that Apoe−/−Ido−/− mice presented no overt alteration in plasma lipids under a chow diet [10], suggesting that the strain background could play a major role in how mice respond to variations in IDO1-mediated Trp metabolism. Considering that the kynurenine pathway has been implicated in the regulation of SREBP-2 [30], which, in addition to regulating cholesterol synthesis, also regulates LDLR expression, it seems plausible that the presence of LDL-receptor in the model could have implications to the degree of hepatic lipid accumulation [47].
As expected, we found that the Kyn/Trp ratio was decreased in the arteries and plasma from Apoe−/−Ido1−/− mice, compared to Apoe−/− mice. Surprisingly, the Kyn/Trp ratio in the liver of both groups was not different at the end of the experiment, suggesting that compensatory mechanisms might have been triggered in Apoe−/−Ido1−/− mice under HFCD or NASH that could maintain Trp degradation rates. In the context of human liver disease, it has been shown that high Kyn/Trp ratio is associated with greater liver fibrosis in the context of HIV and HCV infections, as well as in patients with acute decompensation and acute-on-chronic liver failure cirrhosis [48,49]. Considering that in the current work, after 7.0 weeks of HFCD, our model developed just early stages of liver disease and mild fibrosis, we can speculate that worsening of NASH could lead to altered kynurenine pathway metabolism, which needs to be validated in future studies. In line with the previous thought, it has been shown that kynurenine pathway activity was found to be normal in patients with compensated cirrhosis, and only changed with aggravation of the disease [49].
While IDO1 is thought to be an inducible enzyme triggered especially by proinflammatory factors such as interferon-γ (IFNγ), Trp can also be degraded by two other enzymes, the IDO1 paralogues IDO2 and TDO2. It has been suggested that TDO2 is constitutively expressed in the brain and in the liver. While some regulation redundancy/overlap between IDO1 and IDO2 expression has been suggested, it has been thought that TDO2 expression is mainly mediated by glucocorticoids and other hormones [29]. In our study, we observed that TDO2 is upregulated in the liver of Apoe−/−Ido1−/−, as well as Apoe−/− mice over time on diet, suggesting that alterations in lipids and/or inflammation, known to be induced in hyperlipidaemic mice over time [50], could regulate hepatic TDO2 regulation. Hence, the fact that TDO2 protein expression in the aortas do not follow the same pattern suggests that this enzyme plays a rather liver-specific role.
It has been shown in a murine model of liver fibrosis with CCL4 that hepatic Tdo2 is upregulated in Ido1−/− mice [18]. In this study, the authors showed that Tdo2 upregulation was associated with increased expression of the general control nonderepressive-2 kinase (GCN2), a key nutrient sensor that is also known to be regulated by changes in amino acid metabolism [51]. As mentioned earlier, TDO2 is a haemoprotein that requires Fe2+ for its full activation. Hence, it has been proposed that haem promotes the de novo synthesis of TDO2, which constitutes an important mechanism of regulation of Trp degradation by this enzyme [33]. Using HepG2 hepatoma cultures, we found that excess fatty acids significantly downregulated, while iron upregulated TDO2 mRNA levels. Considering that lipid and iron accumulation are common features of NAFLD/NASH progression, our data suggest that these ‘nutrients’ could be involved in the transcription regulation of hepatic TDO2, regulation of Trp metabolism, as well as control of hepatic inflammation.
As previously mentioned, IDO1-mediated immunoregulatory mechanisms could be the consequence of Trp depletion or the production of bioactive metabolites. In this context, there are bulk data indicating that kynurenines can influence immune responses in a paracrine fashion, e.g., the overexpression of IDO1 by tumours increases the production of L-Kyn and 3-HAA that can signal to inhibit effector T-cell responses or promote Treg differentiation [52,53]; the latter two outcomes have been recognized as an important mechanism of immune escape by tumours. Using conditioned media from HepG2 cells that were treated with palmitic acid or iron, on THP-1 macrophages, indicated that the regulation of TDO2 expression and Trp hepatic catabolism, through the kynurenine pathway, could constitute an important mechanism of communication between hepatocytes and macrophages, and the development of liver inflammation, particularly driven by IL-1β.
Surprisingly for us, Tdo2 expression increased over time, not only in the livers of Apoe−/− Ido1−/− but also in Apoe−/− mice fed HFCD, which can express Ido1; however, the upregulation of Tdo2 in the latter strain did not result in increased hepatic Trp degradation. These results raise two major thoughts: first, that TDO2-associated Trp metabolism and its potential influence on liver inflammation is independent of IDO1; and second, that in the context of NAFLD/NASH, hepatic Trp levels and metabolism is more complex than we anticipated. The uptake of Trp is thought to be driven essentially by the L-type neutral amino acid transporter 1 (LAT1 or Slc7a5). In this context, it has been shown that LPS and TNF significantly reduce LAT1 and Trp uptake in neuron-like cells [54], while IL-1β upregulates LAT1 levels in fibroblast-like synoviocytes [55]. These findings suggest that LAT1 is an interesting candidate for future research involving Trp metabolism in the context of liver disease. Notably, the uptake of Trp can be regulated due to competition with other amino acids, and alterations in the levels and metabolism of amino acids, besides Trp, have been associated with NAFLD, e.g., the other aromatic amino acids tyrosine and phenylalanine, arginine, and branched-chain amino acids [56,57]. However, less is known about these other amino acids and their role in the regulation of immunometabolic responses, warranting further investigation.
In conclusion, despite the small size and short study design, we demonstrate that Apoe−/− mice fed HFCD for 7 weeks is a plausible model to study liver disease with atherosclerosis as a major comorbidity. Evaluation of the effects of Ido1 genetic ablation revealed that this model may be used to better understand the dichotomies between vascular and hepatic inflammatory processes. A complete understanding of the role of IDO1 in the modulation of cardiovascular and liver disease as a comorbidity warrants further investigations. These could include the compensatory effects of other kynurenine pathway enzymes, and the potential crosstalk between hepatic and immune cells mediated by different Trp metabolites. A better understanding of these molecular processes could have implications for the design of high-precision therapies that can benefit both atherosclerosis and NAFLD/NASH.
4. Methods
4.1. Animal Model
The Apoe−/−Ido1−/− mouse strain was generated by crossing Apoe−/− mice with Ido1−/− mice at the Kennedy Institute of Rheumatology, Oxford, UK [10]. The strain was transferred to the Center for Molecular Medicine at the Karolinska Institute in Stockholm and bred with Apoe−/− (B6.129P2-Apoetm1Unc/J, strain code 622, JAX™, Charles River, The Netherlands) to generate Apoe−/− and Apoe−/−Ido1−/− littermate control mice that were used in the study; all mice were kept in specific pathogen-free (SPF) conditions with a 12-h light/dark cycle throughout the study. The model of concomitant development of atherosclerosis and NASH was achieved by adapting the protocol from Schierwagen et al. (2015) [36]. Briefly, 10-week-old male mice were fed normal chow or a high-fat, cholesterol-rich diet (HFCD) containing 42% kcal from fat, 43% kcal from carbohydrate, 15% kcal from protein, and 1.25% cholesterol (E15723-34, Sniff, Germany) ad libitum for 3.5 or 7 weeks. All animal experiments were performed in accordance with national guidelines and approved by the Stockholm Norra Regional Ethics Board (N28-15, approved on 26 March 2015), which conforms to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
4.2. Atherosclerosis Burden Analyses
At the end of treatment, mice were euthanized with CO2. Blood was collected by cardiac puncture, and vascular perfusion was performed with sterile RNase-free PBS. After perfusion, the heart and aortic arch were dissected and preserved for lesion and immunohistochemistry analyses. We have previously shown that Ido1 genetic and pharmacological ablation increases plaque burden in the aortic root [9,10]. In this study, aortic root sections obtained from cryopreserved hearts were used to obtain representative micrographs of plaque burden, which confirmed our previous publications. Lesion size was visualized on haematoxylin- and oil red O-stained sections as previously described [58]. Macrophage content in plaques was visualized using primary antibodies against Mac-2 (Cedarlane Laboratories, Burlington, ON, Canada) that were applied to acetone-fixed cryosections. Detection was performed using an ABC alkaline phosphatase kit (Vector Laboratories, Burlingame, CA, USA) as previously described [59]. En face lipid accumulation in the mouse aortic arch was determined using Sudan IV staining. Images were captured using a Leica DC480 camera connected to a Leica MZ6 stereomicroscope (Leica, Wetzlar, Germany). The lesion area was calculated using ImageJ software (NIH, Bethesda, MD, USA). Samples that were damaged during processing or analysis were excluded from the study. For the assessment of plaques, samples were coded, and the evaluation was performed by trained personnel who were blinded to the treatment groups.
4.3. Histological Analysis of Liver Disease Burden
The livers were dissected, and analogous samples were either snap-frozen or fixed in 4% phosphate-buffered formaldehyde for histopathology analyses. After fixation for 24–48 h, samples were dehydrated in a series of graded alcohols and embedded in paraffin wax. Serial sections of 5 μm were rehydrated and subjected to haematoxylin and eosin staining for morphological visualization of liver damage, and Picrosirius red (Fluka-Sigma Aldrich, Switzerland) to evaluate the extent of fibrosis. Hepatic macrophage content was evaluated using a primary antibody against Mac-2 (Cedarlane Laboratories, Burlington, Canada) that was detected using goat anti-rat IgG (DyLight® 594) as the secondary antibody (Abcam, Cambridge, UK), and nuclei were stained with DAPI (Sigma Aldrich, St. Louis, MO, USA). All histological assessments were performed by a trained examiner who was blinded to the groups.
4.4. Biochemical Parameters in Liver and Blood
Analogous segments of snap-frozen livers were lysed in RIPA buffer using a TissueLyser II (Qiagen, Germantown, MD, USA). Hepatic hydroxyproline content was evaluated using a colorimetric assay kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Liver lipids were extracted from liver samples using the Folch method [60]. Briefly, lysates were homogenized in methanol, and lipids were extracted by chloroform separation (methanol: chloroform (1:2)). After drying, the extracts were redissolved in 1% Triton-100, and cholesterol and triglyceride contents were measured using enzymatic colorimetric kits (Randox Lab. Ltd. Crumlin, UK) according to the manufacturer’s instructions. Biochemical parameters (alanine aminotransferase (ALT) and aspartate aminotransferase (AST)) in blood were evaluated on a Samsung PT10V clinical chemistry analyser. Plasma cholesterol and triglycerides were measured using enzymatic colorimetric kits (Randox Lab. Ltd., Crumlin, UK) according to the manufacturer’s instructions. Hepatic free iron content was determined using a colorimetric assay kit (Sigma-Aldrich, St. Louis, MO, USA) following the manufacturer’s instructions.
4.5. Evaluation of Inflammatory Markers
In addition to immunohistological analyses, inflammation was evaluated in liver samples by qPCR. RNA was isolated from mouse livers using a RNeasy kit (Qiagen, Hilden, Germany). After approving the quality of the RNA on a NanoDrop (Thermo Scientific, Waltham, MA, USA), it was reverse transcribed with a High-Capacity RNA-to-cDNA™ Kit (Thermo Scientific, Waltham, MA, USA ) and amplified by real-time PCR using assay-on-demand primers and probes (Il12, Cd80, Cxcl10, Chil3, Arg1, Cd206, Tdo2, TDO2; all from Thermo Scientific, Waltham, MA, USA) in an ABI 7700 Sequence Detector (Applied Biosystems, Foster City, CA, USA). Hypoxanthine guanidine ribonucleosyl transferase (HPRT) was used as a housekeeping gene. Assay-on-demand primers and probes are provided in Supplementary Table S1. Data were analysed based on the relative expression method with the formula 2−ΔΔCT, where ΔΔCT = ΔCT (sample)–ΔCT (calibrator = average CT values of all samples within the control group) and ΔCT is the average CT of the housekeeping genes subtracted from the CT of the target gene. The levels of cytokines, including TNF- α, IL-1β, CCL2, and IL-10, were measured by ELISA according to the manufacturer’s instructions (all from R&D Systems, Minneapolis, MN, USA).
4.6. HepG2 Culture and Treatments
The human hepatoma cell line HepG2 was purchased from ATCC (VA, USA) and cultured as previously described [30]. Briefly, cells were maintained in low glucose (1 g/L) Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% foetal bovine serum (FBS), 2 mM L-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin (all from Gibco, UK). At least 14 days prior to the experiment, the cells were passaged, and the medium was replaced with low glucose (1 g/L) DMEM supplemented with 2% AB+ human serum (Blodcentralen Karolinska Universitetssjukhuset, Sweden), 2 mM L-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin. For mRNA analysis, cells were treated for 24 h with 500 μM palmitic acid (Sigma-Aldrich, St. Louis, MO, USA), 100 μM FeSO4 (Sigma Aldrich, St. Louis, MO, USA), 0.62 μM TDO inhibitor LM10 (Sigma-Aldrich, St. Louis, MO, USA) in different combinations, or vehicle as detailed in the figure legends. In parallel experiments, cells were washed after 24 h of treatment with palmitic acid or FeSO4, and the TDO inhibitor LM10 (all from Sigma-Aldrich, St. Louis, MO, USA), and new media was added. Supernatants of these cultures were saved after 48 h and used as conditional media in THP-1 cultures.
4.7. IL-1β Secretion by THP-1 Macrophages Treated with HepG2-Conditioned Media
The human monocytic cell line THP-1 was maintained in culture using RPMI 1640 (Invitrogen, MA, USA) culture medium containing 10% heat-inactivated FBS (Gibco, UK) supplemented with 2 mM L-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin (all from Gibco, UK). THP-1 monocytes were then differentiated into macrophages by 24 h incubation with 100 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, St. Louis, MO, USA), followed by 24 h incubation with conditioned media from HepG2 cells subjected to different treatments and upon stimulation with 10 ng/mL LPS (Sigma-Aldrich, St. Louis, MO, USA) for 4 h. After 4 h of incubation with LPS, cells were treated with 5 μM ATP for inflammasome activation and the release of IL-1β was measured by ELISA as previously described.
4.8. Kyn/Trp Ratio
The L-Kyn/Trp ratio, determined by ELISA (ImmuSmol, Bordeaux France), was used as a surrogate marker of IDO1-TDO2 activity in the aorta, liver, and plasma from 7-week HFCD-fed Apoe−/− and Apoe−/−Ido1−/− mice.
4.9. Statistical Analysis
The results are presented as the mean ± SEM if not otherwise stated. The Mann–Whitney U-test was used for comparisons between two groups, and Kruskal–Wallis ANOVA with Dunn’s post-test was used for comparisons between more than two groups. Correlations were calculated using simple linear regression analysis. p values < 0.05 were considered significant.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23095203/s1.
Author Contributions
Conceptualization, A.A., G.L.T., I.K. and D.F.J.K.; methodology, A.A., G.L.T., I.K. and D.F.J.K.; formal analysis, A.A. and G.L.T.; investigation, A.A. and G.L.T., with support from M.B., M.J.F., A.G., S.G. and F.B.C.; resources, J.C., C.M. and D.F.J.K.; data curation, A.A. and G.L.T.; writing—original draft preparation, A.A., G.L.T. and D.F.J.K.; writing—review and editing, A.A., G.L.T., J.O.M., C.M. and D.F.J.K.; supervision, J.O.M., D.S.P.A., C.M. and D.F.J.K.; project administration, D.F.J.K.; funding acquisition, G.L.T., D.S.P.A., F.B.C., J.O.M. and D.F.J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Swedish Heart-Lung Foundation, the Novo Nordisk Foundation (MSAM consortium, NNF15SA0018346; MeRIAD consortium, 0064142), and the University of Southern Denmark. F.B.C. was supported by the foundation ‘Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil’ (CAPES; Finance Code 001). G.L.T. was supported by ‘The São Paulo Research Foundation’ (FAPESP; 2019/13598-8). J.O.M. was supported by FAPESP (2020/03175-0).
Institutional Review Board Statement
The animal study protocol was approved by the Stockholm Norra Regional Ethics Board (N28-15, approved on 26 March 2015).
Informed Consent Statement
Not applicable.
Data Availability Statement
Available on request from the corresponding author.
Acknowledgments
We thank Anneli Olsson and Linda Haglund for their technical assistance.
Conflicts of Interest
I.K. reports personal fees from Orion Pharma unrelated to the submitted work. The remaining authors declare no conflict of interest.
References
- Nichols, M.; Townsend, N.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe 2014: Epidemiological update. Eur. Heart J. 2014, 35, 2950–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Health Risks: Mortality and Burden of Disease Attributable to Selected Major Risks; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- Cholesterol Treatment Trialists, Collaborators; Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Ketelhuth, D.F.; Hansson, G.K. Adaptive Response of T and B Cells in Atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunon, J.; Back, M.; Badimon, L.; Bochaton-Piallat, M.L.; Cariou, B.; Daemen, M.J.; Egido, J.; Evans, P.C.; Francis, S.E.; Ketelhuth, D.F.; et al. Interplay between hypercholesterolaemia and inflammation in atherosclerosis: Translating experimental targets into clinical practice. Eur. J. Prev. Cardiol. 2018, 25, 948–955. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Ibrahim, M.; Singh, C.; Ganie, M.A.; Alsayari, K. NASH: The Hepatic injury of Metabolic syndrome: A brief update. Int. J. Health Sci. (Qassim) 2009, 3, 265–270. [Google Scholar]
- Ketelhuth, D.F.J. The immunometabolic role of indoleamine 2,3-dioxygenase in atherosclerotic cardiovascular disease: Immune homeostatic mechanisms in the artery wall. Cardiovasc. Res. 2019, 115, 1408–1415. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, K.A.; Ovchinnikova, O.; Berg, M.; Baumgartner, R.; Agardh, H.; Pirault, J.; Gisterå, A.; Assinger, A.; Laguna-Fernandez, A.; Bäck, M.; et al. Inhibition of indoleamine 2,3-dioxygenase promotes vascular inflammation and increases atherosclerosis in Apoe−/− mice. Cardiovasc. Res. 2015, 106, 295–302. [Google Scholar] [CrossRef]
- Cole, J.E.; Astola, N.; Cribbs, A.P.; Goddard, M.E.; Park, I.; Green, P.; Davies, A.H.; Williams, R.O.; Feldmann, M.; Monaco, C. Indoleamine 2,3-dioxygenase-1 is protective in atherosclerosis and its metabolites provide new opportunities for drug development. Proc. Natl. Acad. Sci. USA 2015, 112, 13033–13038. [Google Scholar] [CrossRef] [Green Version]
- Daissormont, I.; Christ, A.; Temmerman, L.; Millares, S.; Seijkens, T.; Manca, M.; Rousch, M.; Poggi, M.; Boon, L.; van der Loos, C.; et al. Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-Cell proliferation and activity novelty and significance. Circ. Res. 2011, 109, 1387–1395. [Google Scholar] [CrossRef]
- Yun, T.J.; Lee, J.S.; Machmach, K.; Shim, D.; Choi, J.; Wi, Y.J.; Jang, H.S.; Jung, I.H.; Kim, K.; Yoon, W.K.; et al. Indoleamine 2,3-Dioxygenase-expressing aortic plasmacytoid dendritic cells protect against atherosclerosis by induction of regulatory T Cells. Cell Metab. 2016, 24, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuffy, M.C.; Silverio, A.M.; Qin, L.; Wang, Y.; Eid, R.; Brandacher, G.; Lakkis, F.G.; Fuchs, D.; Pober, J.S.; Tellides, G. Induction of indoleamine 2,3-dioxygenase in vascular smooth muscle cells by interferon-gamma contributes to medial immunoprivilege. J. Immunol. 2007, 179, 5246–5254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metghalchi, S.; Ponnuswamy, P.; Simon, T.; Haddad, Y.; Laurans, L.; Clément, M.; Dalloz, M.; Romain, M.; Esposito, B.; Koropoulis, V.; et al. Indoleamine 2,3-dioxygenase fine-tunes immune homeostasis in atherosclerosis and colitis through repression of interleukin-10 production. Cell Metab. 2015, 22, 460–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, R.; Berg, M.; Matic, L.; Polyzos, K.P.; Forteza, M.J.; Hjorth, S.A.; Schwartz, T.W.; Paulsson-Berne, G.; Hansson, G.K.; Hedin, U.; et al. Evidence that a deviation in the kynurenine pathway aggravates atherosclerotic disease in humans. J. Intern. Med. 2021, 289, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kwok, M.K.; Fong, S.S.M.; Schooling, C.M. Indoleamine 2,3-dioxygenase and ischemic heart disease: A Mendelian Randomization study. Sci. Rep. 2019, 9, 8491. [Google Scholar] [CrossRef] [Green Version]
- Nagano, J.; Shimizu, M.; Hara, T.; Shirakami, Y.; Kochi, T.; Nakamura, N.; Ohtaki, H.; Ito, H.; Tanaka, T.; Tsurumi, H.; et al. Effects of indoleamine 2,3-dioxygenase deficiency on high-fat diet-induced hepatic inflammation. PLoS ONE 2013, 8, e73404. [Google Scholar] [CrossRef]
- Ogiso, H.; Ito, H.; Ando, T.; Arioka, Y.; Kanbe, A.; Ando, K.; Ishikawa, T.; Saito, K.; Hara, A.; Moriwaki, H.; et al. The Deficiency of Indoleamine 2,3-Dioxygenase Aggravates the CCl4-Induced Liver Fibrosis in Mice. PLoS ONE 2016, 11, e0162183. [Google Scholar] [CrossRef] [Green Version]
- Wolowczuk, I.; Hennart, B.; Leloire, A.; Bessede, A.; Soichot, M.; Taront, S.; Caiazzo, R.; Raverdy, V.; Pigeyre, M.; ABOS Consortium; et al. Tryptophan metabolism activation by indoleamine 2,3-dioxygenase in adipose tissue of obese women: An attempt to maintain immune homeostasis and vascular tone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R135–R143. [Google Scholar] [CrossRef] [Green Version]
- Milosavljevic, N.; Gazdic, M.; Simovic Markovic, B.; Arsenijevic, A.; Nurkovic, J.; Dolicanin, Z.; Jovicic, N.; Jeftic, I.; Djonov, V.; Arsenijevic, N.; et al. Mesenchymal stem cells attenuate liver fibrosis by suppressing Th17 cells—An experimental study. Transpl. Int. 2018, 31, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Laurans, L.; Venteclef, N.; Haddad, Y.; Chajadine, M.; Alzaid, F.; Metghalchi, S.; Sovran, B.; Denis, R.G.P.; Dairou, J.; Cardellini, M.; et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat. Med. 2018, 24, 1113–1120. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Stepanova, M.; Younossi, Z.M. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin. Gastroenterol. Hepatol. 2012, 10, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Despres, J.P.; Koh, K.K. Prevention of atherosclerosis in overweight/obese patients. In need of novel multi-targeted approaches. Circ. J. 2011, 75, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, J.J.; Beckman, J.A.; Jaffe, I.Z. Sex as a Biological Variable in Atherosclerosis. Circ. Res. 2020, 126, 1297–1319. [Google Scholar] [CrossRef] [PubMed]
- Criado, G.; Simelyte, E.; Inglis, J.J.; Essex, D.; Williams, R.O. Indoleamine 2,3 dioxygenase-mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen-induced arthritis. Arthritis Rheum. 2009, 60, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Ciorba, M.A. Indoleamine 2,3 dioxygenase in intestinal disease. Curr. Opin. Gastroenterol. 2013, 29, 146–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Berg, M.; Polyzos, K.A.; Agardh, H.; Baumgartner, R.; Forteza, M.J.; Kareinen, I.; Gistera, A.; Bottcher, G.; Hurt-Camejo, E.; Hansson, G.K.; et al. 3-Hydroxyanthralinic acid metabolism controls the hepatic SREBP/lipoprotein axis, inhibits inflammasome activation in macrophages, and decreases atherosclerosis in Ldlr-/- mice. Cardiovasc. Res. 2020, 116, 1948–1957. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Takei, Y. Iron overload in nonalcoholic steatohepatitis. Adv. Clin. Chem. 2011, 55, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Correia, M.A. Heme: A regulator of rat hepatic tryptophan 2,3-dioxygenase? Arch. Biochem. Biophys. 2000, 377, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- von Scheidt, M.; Zhao, Y.; Kurt, Z.; Pan, C.; Zeng, L.; Yang, X.; Schunkert, H.; Lusis, A.J. Applications and Limitations of Mouse Models for Understanding Human Atherosclerosis. Cell Metab. 2017, 25, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Schierwagen, R.; Maybuchen, L.; Zimmer, S.; Hittatiya, K.; Back, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef]
- Bieghs, V.; Van Gorp, P.J.; Wouters, K.; Hendrikx, T.; Gijbels, M.J.; van Bilsen, M.; Bakker, J.; Binder, C.J.; Lutjohann, D.; Staels, B.; et al. LDL receptor knock-out mice are a physiological model particularly vulnerable to study the onset of inflammation in non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e30668. [Google Scholar] [CrossRef] [Green Version]
- van den Hoek, A.M.; Verschuren, L.; Worms, N.; van Nieuwkoop, A.; de Ruiter, C.; Attema, J.; Menke, A.L.; Caspers, M.P.M.; Radhakrishnan, S.; Salic, K.; et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells 2020, 9, 2014. [Google Scholar] [CrossRef]
- Bedossa, P. Pathology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. 1), 85–89. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef]
- Polyzos, K.A.; Ketelhuth, D.F. The role of the kynurenine pathway of tryptophan metabolism in cardiovascular disease. An emerging field. Hamostaseologie 2015, 35, 128–136. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.P.; Hahn, M. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of indoleamine 2,3-dioxygenase expression and kynurenine pathway metabolites levels in serum samples of diabetic retinopathy patients. Arch. Physiol. Biochem. 2011, 117, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H. Regulation of proinsulin synthesis in pancreatic islets and a new aspect to insulin-dependent diabetes. Mol. Cell. Biochem. 1981, 37, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Kotake, Y.; Kotake, Y. Studies on the urinary excretion of xanthurenic acid in diabetics. Acta Vitaminol. Et Enzymol. 1984, 6, 221–228. [Google Scholar]
- Zhang, L.; Ovchinnikova, O.; Berg, M.; Baumgartner, R.; Agardh, H.; Pirault, J.; Gistera, A.; Assinger, A.; Laguna-Fernandez, A.; Back, M.; et al. The tryptophan metabolite 3-hydroxyanthranilic acid lowers plasma lipids and decreases atherosclerosis in hypercholesterolaemic mice. Eur. Heart J. 2012, 33, 2025–2034. [Google Scholar] [CrossRef]
- Chang, M.Y.; Smith, C.; DuHadaway, J.B.; Pyle, J.R.; Boulden, J.; Soler, A.P.; Muller, A.J.; Laury-Kleintop, L.D.; Prendergast, G.C. Cardiac and gastrointestinal liabilities caused by deficiency in the immune modulatory enzyme indoleamine 2,3-dioxygenase. Cancer Biol. Ther. 2011, 12, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Claria, J.; Moreau, R.; Fenaille, F.; Amoros, A.; Junot, C.; Gronbaek, H.; Coenraad, M.J.; Pruvost, A.; Ghettas, A.; Chu-Van, E.; et al. Orchestration of Tryptophan-Kynurenine Pathway, Acute Decompensation, and Acute-on-Chronic Liver Failure in Cirrhosis. Hepatology 2019, 69, 1686–1701. [Google Scholar] [CrossRef]
- Kardashian, A.; Ma, Y.; Yin, M.T.; Scherzer, R.; Nolan, O.; Aweeka, F.; Tien, P.C.; Price, J.C. High Kynurenine:Tryptophan Ratio Is Associated With Liver Fibrosis in HIV-Monoinfected and HIV/Hepatitis C Virus-Coinfected Women. Open Forum Infect. Dis. 2019, 6, ofz281. [Google Scholar] [CrossRef]
- Meir, K.S.; Leitersdorf, E. Atherosclerosis in the apolipoprotein-E-deficient mouse: A decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Battu, S.; Minhas, G.; Mishra, A.; Khan, N. Amino Acid Sensing via General Control Nonderepressible-2 Kinase and Immunological Programming. Front. Immunol. 2017, 8, 1719. [Google Scholar] [CrossRef] [Green Version]
- Hornyak, L.; Dobos, N.; Koncz, G.; Karanyi, Z.; Pall, D.; Szabo, Z.; Halmos, G.; Szekvolgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyawali, A.; Kang, Y.S. Pretreatment Effect of Inflammatory Stimuli and Characteristics of Tryptophan Transport on Brain Capillary Endothelial (TR-BBB) and Motor Neuron Like (NSC-34) Cell Lines. Biomedicines 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jiang, C.; Cai, Y.; Guo, Y.; Wang, X.; Zhang, J.; Xu, J.; Xu, K.; Zhu, W.; Wang, S.; et al. Intervening upregulated SLC7A5 could mitigate inflammatory mediator by mTOR-P70S6K signal in rheumatoid arthritis synoviocytes. Arthritis Res. Ther. 2020, 22, 200. [Google Scholar] [CrossRef]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Tome, D. Amino acid metabolism and signalling pathways: Potential targets in the control of infection and immunity. Eur. J. Clin. Nutr. 2021, 75, 1319–1327. [Google Scholar] [CrossRef]
- Centa, M.; Ketelhuth DF, J.; Malin, S.; Gistera, A. Quantification of Atherosclerosis in Mice. J. Vis. Exp. 2019, 148, e59828. [Google Scholar] [CrossRef]
- Gistera, A.; Ketelhuth, D.F. Immunostaining of Lymphocytes in Mouse Atherosclerotic Plaque. Methods Mol. Biol. 2015, 1339, 149–159. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).