
Comparative Transcriptome Analysis Provides Insights into the Resistance in Pueraria [Pueraria lobata (Willd.) Ohwi] in Response to Pseudo-Rust Disease

 , ,
, ,
Abstract
:1. Introduction
2. Results
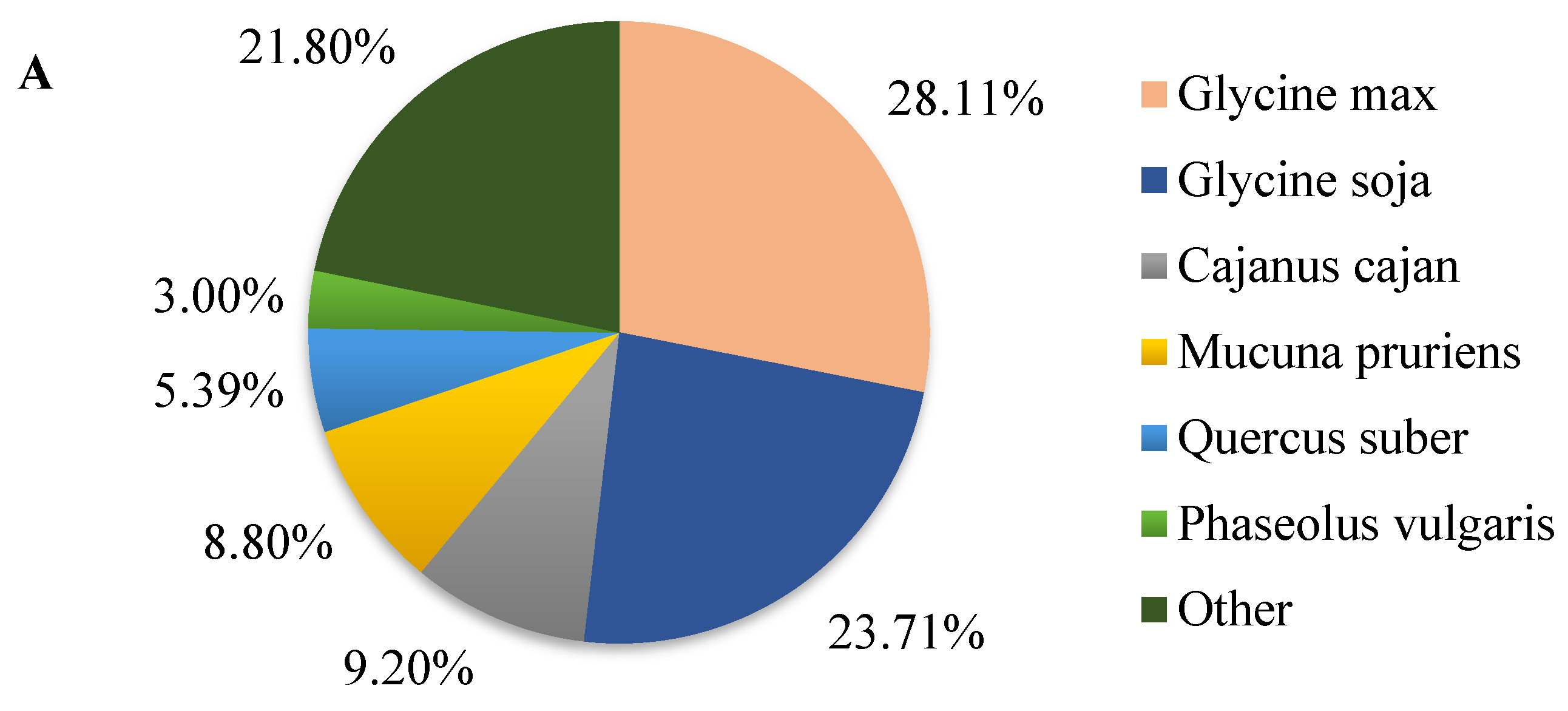
2.1. Transcriptome Analysis
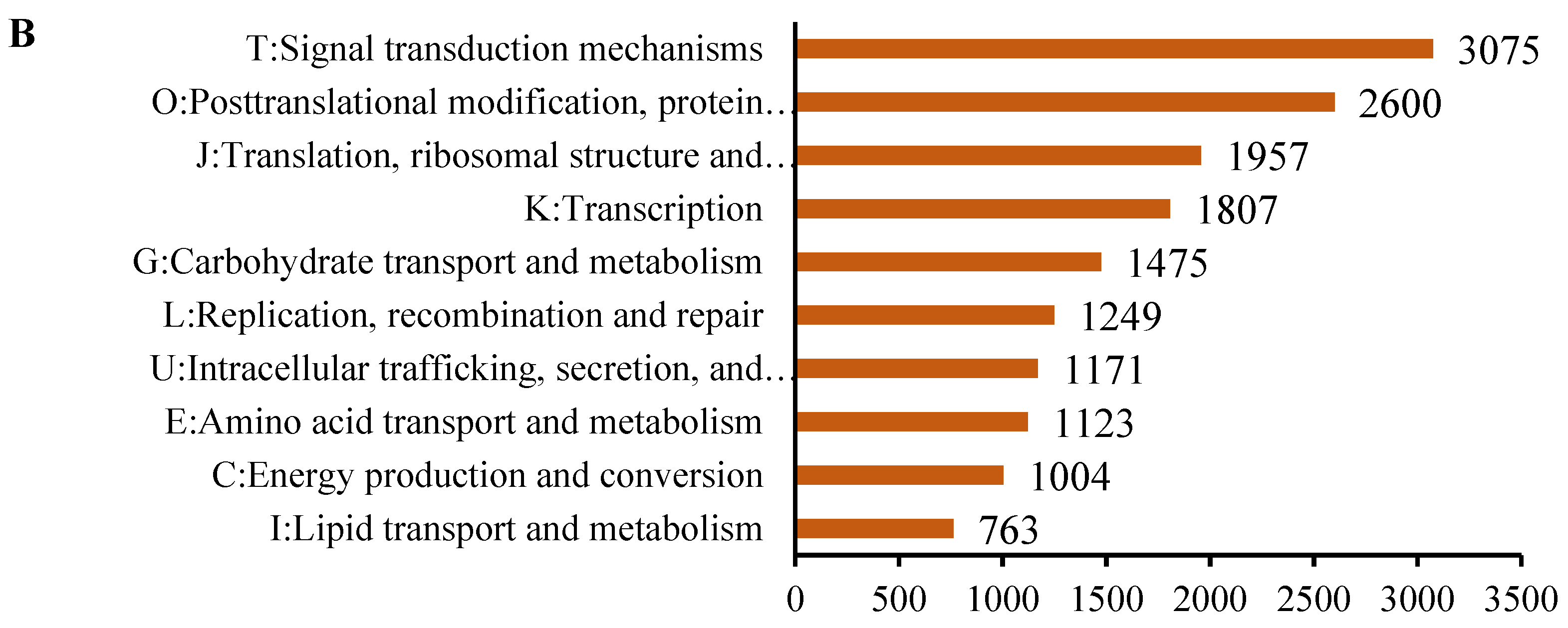
2.2. Overview of Functional Annotation
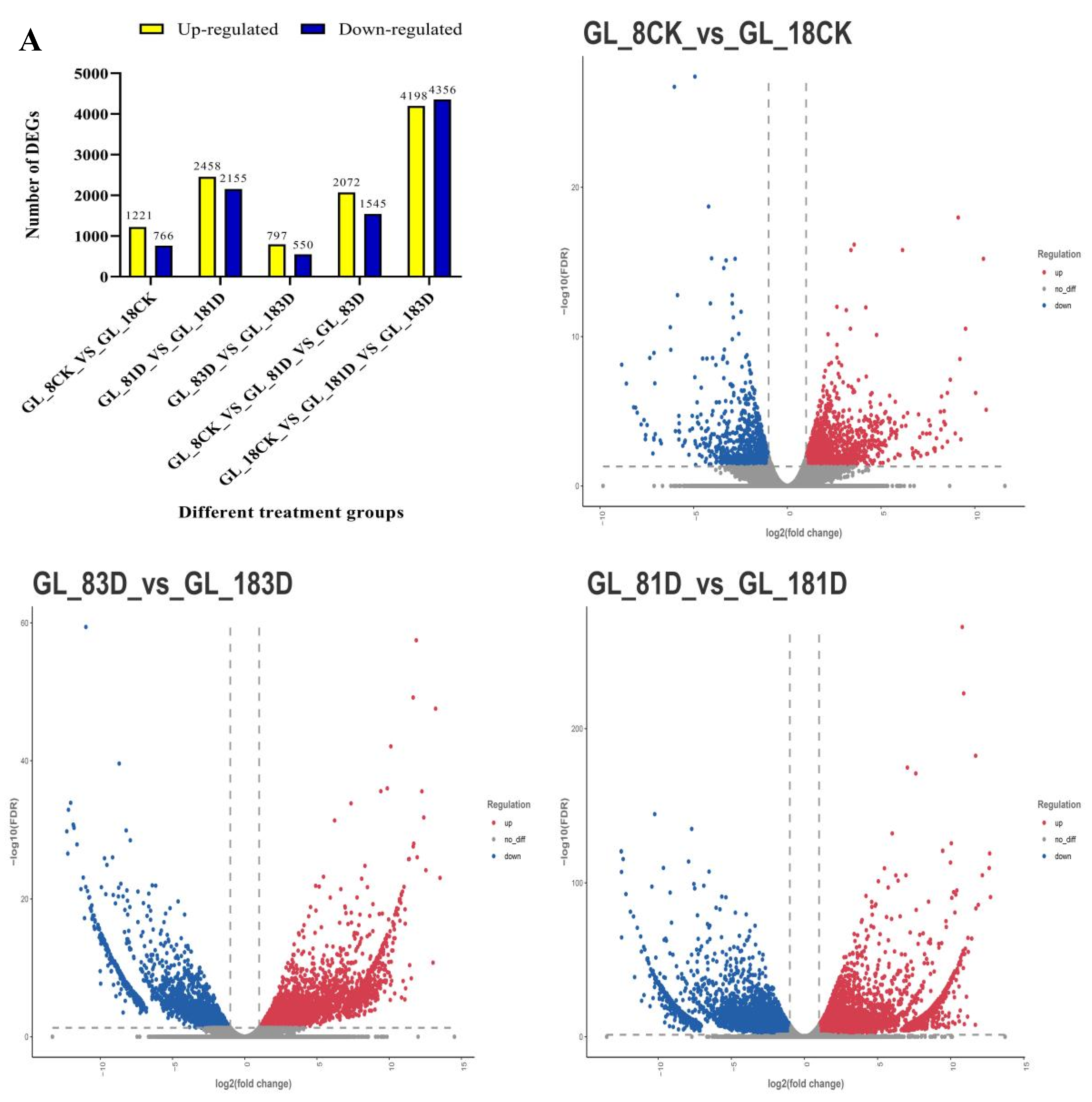
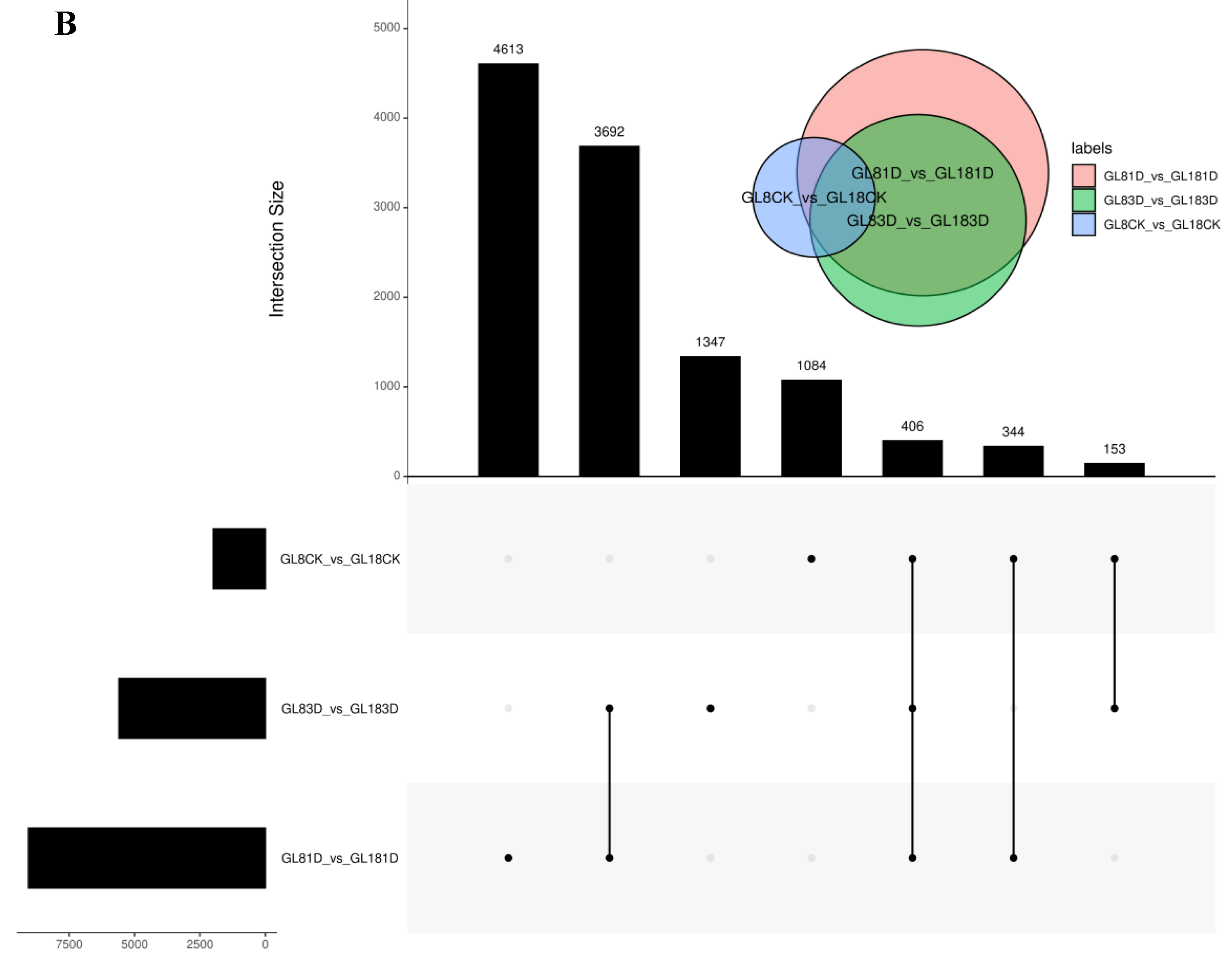
2.3. RNA-Seq Analysis of the Two Cultivars at the Early Stage after SpM Infection and Identification of Differentially Expressed Genes (DEGs)
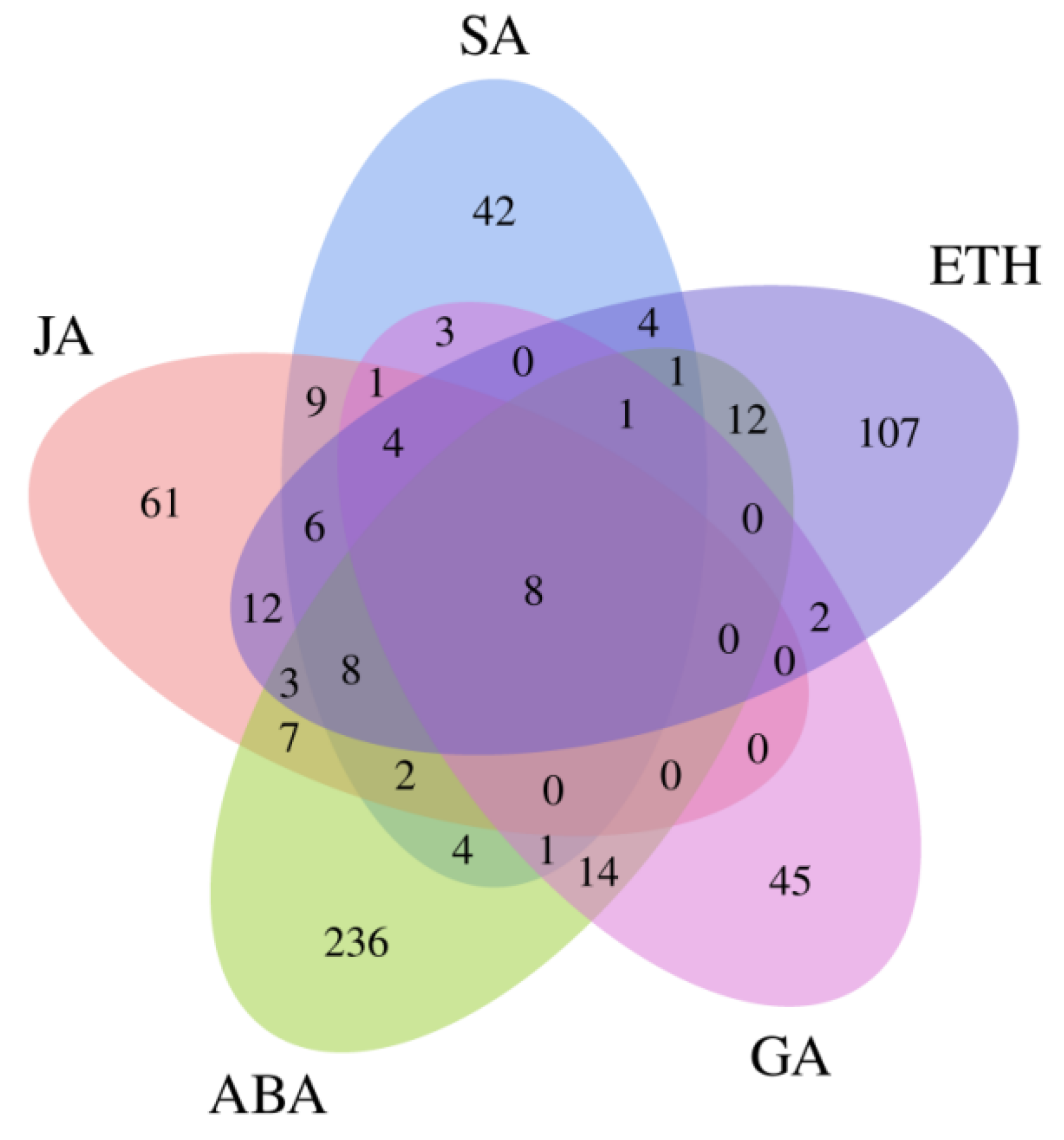
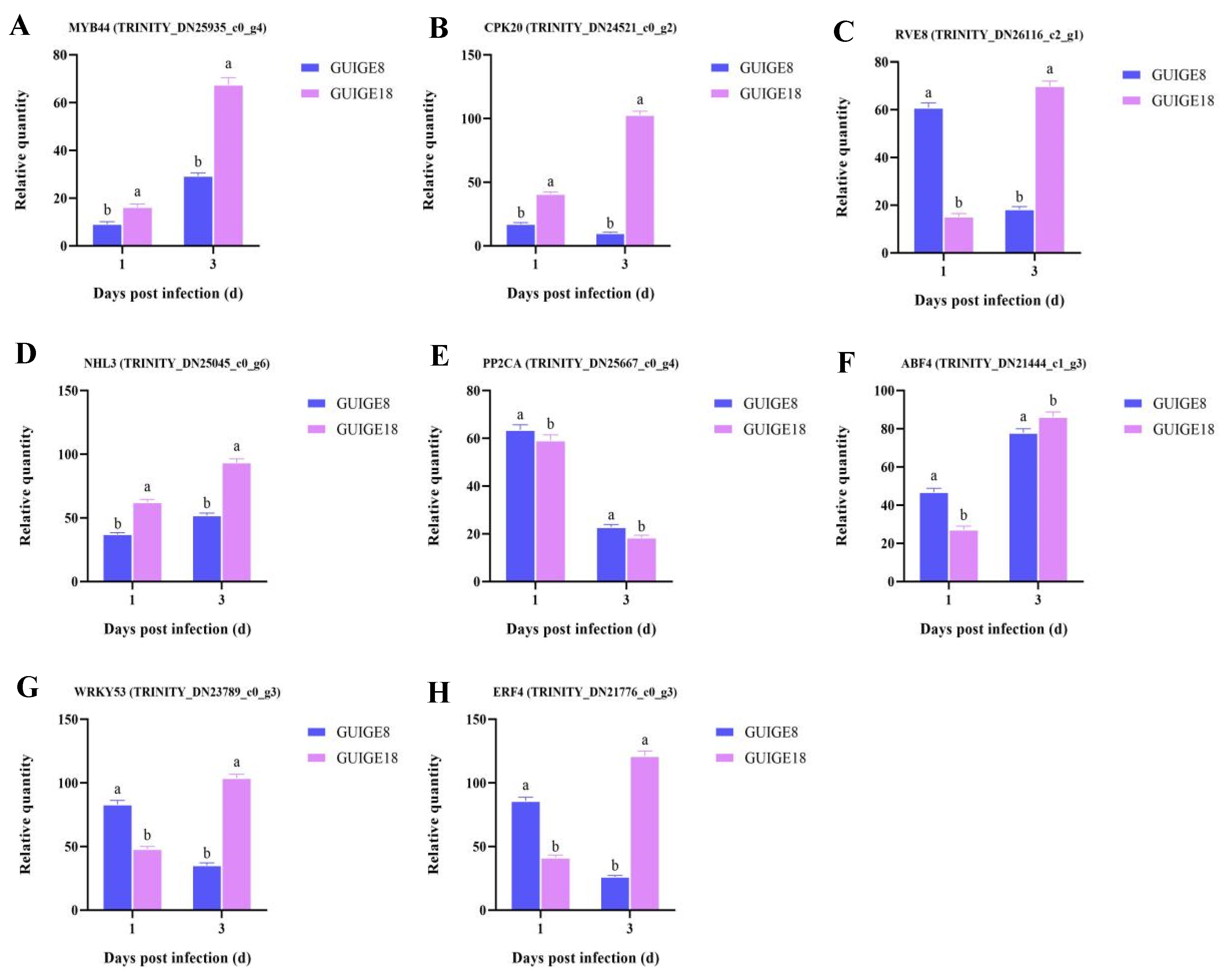
2.4. GO Function Enrichment Analysis of DEGs
2.5. KEGG Pathway Analysis of DEGs
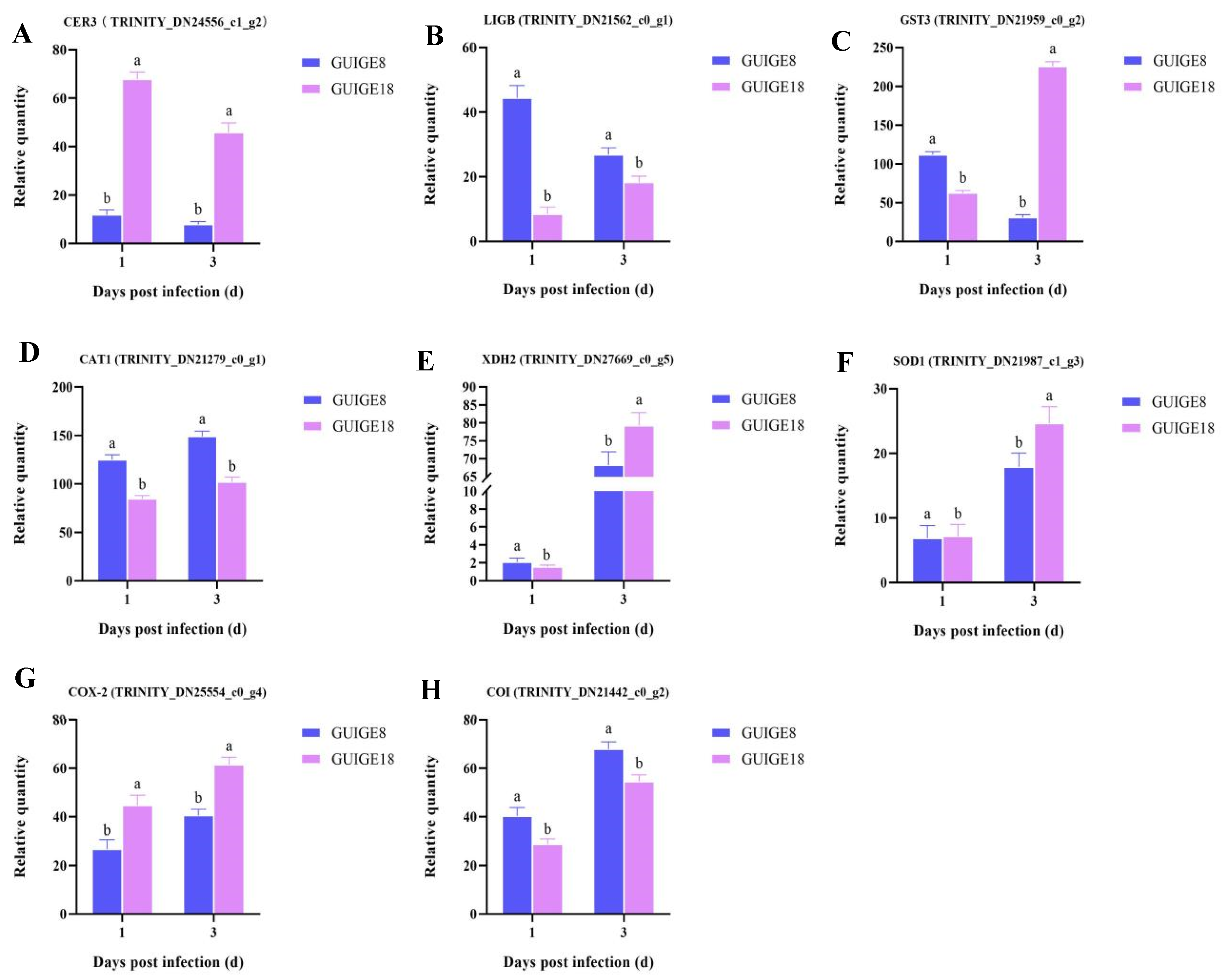
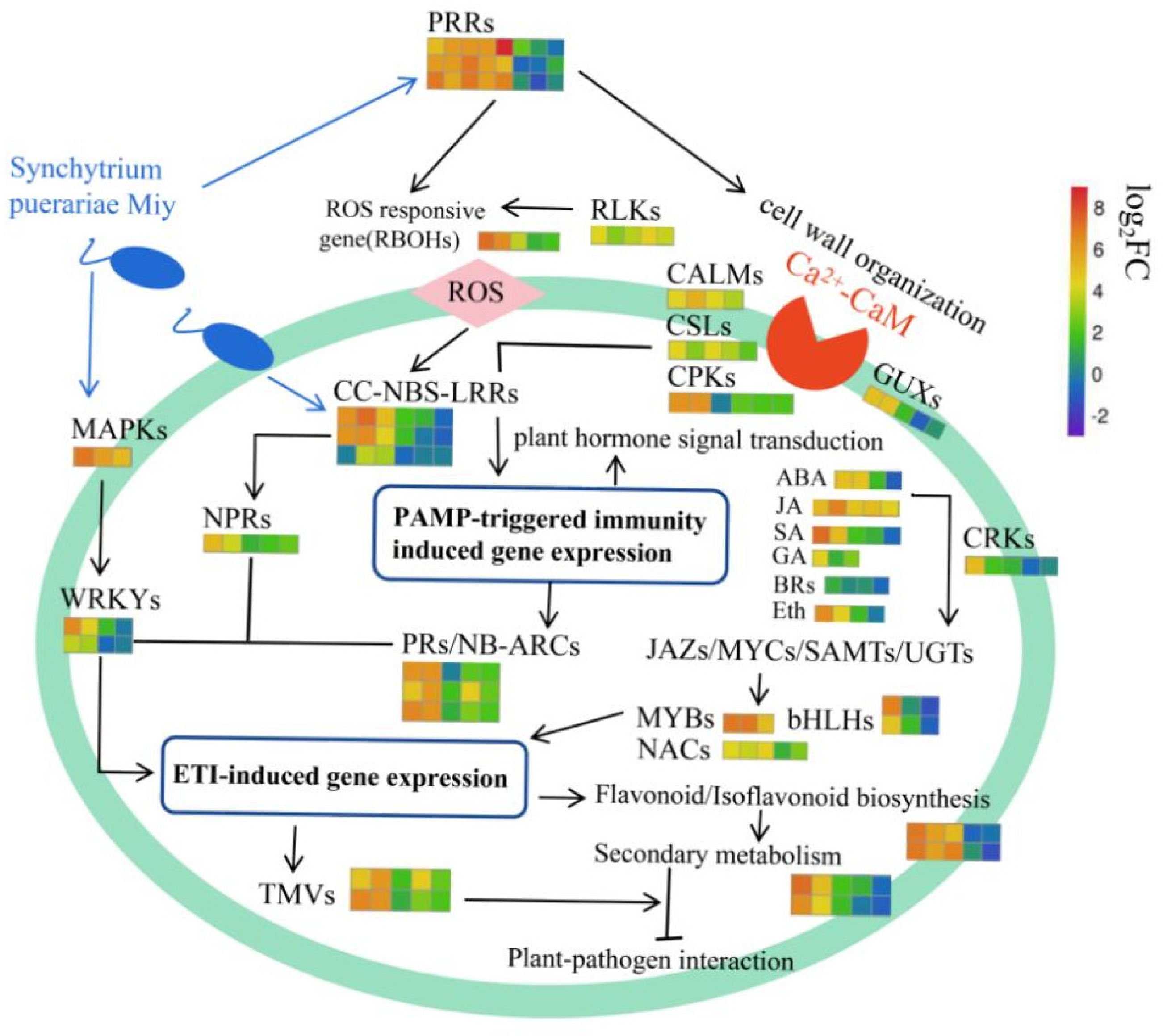
2.6. DEGs Related to Oxidation-Reduction Process
2.7. DEGs Related to Starch and Source Metabolism
2.8. Endogenous Hormone Content and Related Gene Expression
2.9. Flavonoid Content and Related Gene Expression in Flavonoid Biosynthetic Process
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Pathogen Infection
4.2. RNA Extraction, Library Construction, and RNA Sequencing
4.3. De Novo Assembly, Unigene Annotation, and Functional Classification
4.4. Sequencing Analysis and Differential Expression Analysis
4.5. Measurements of Hormones and Defense Enzyme Activity
4.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wing, M.K. Pueraria: The Genus Pueraria; CRC Press: Boca Raton, FL, USA, 2002; ISBN 9781134451883. [Google Scholar]
- Dolly, R.; Visarut, B.; Khwanlada, K.; Wanchai, D.E.; Sornkanok, V. Elicitation of Pueraria candollei var. mirifica suspension cells promises antioxidant potential, implying antiaging activity. Plant Cell Tissue Organ Cult. (PCTOC) 2021, 145, 29–41. [Google Scholar] [CrossRef]
- Wang, S.G.; Zhang, S.M.; Wang, S.P.; Gao, P.; Dai, L. A comprehensive review on Pueraria: Insights on its chemistry and medicinal value. Biomed. Pharmacother. 2020, 131, 110734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.S.; Wang, Y.L.; Zhang, W.Y.; Li, K.D.; Luo, X.F.; Cui, Y.L. Puerarin from Pueraria lobata alleviates the symptoms of irritable bowel syndrome-diarrhea. Food Funct. 2021, 12, 2211–2224. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.F.; Li, J.L.; Meng, X.W.; Zhang, P.Z.; Wu, W.T.; Liu, R.H. Research advances in chemical constituents and pharmacological activities of Pueraria genus. China J. Chin. Mater. Med. 2021, 46, 1311–1331. [Google Scholar] [CrossRef]
- Wang, C.K.; Xu, N.G.; Cui, S. Comparative transcriptome analysis of roots, stems, and leaves of Pueraria lobata (Willd.) Ohwi: Identification of genes involved in isoflavonoid biosynthesis. PeerJ 2021, 9, e10885. [Google Scholar] [CrossRef]
- Zha, Q.; Chen, S.; Zhang, J.; Zhang, X.Y.; Ruan, P.J.; Heba, M.A.; Liu, A. A reliable and rapid pharmacokinetic study of pueraria isoflavones using pueraria reference extractive substance in beagle plasma: Application to study of Yufeng Ningxin Tablets. Chin. Herb. Med. 2020, 12, 88–94. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, L.H.; Chen, J. Pathogenic identification of the imitation rust of Pueraria thomsonii in Guangzhou Area. Plant Prot. 1999, 25, 13–15. [Google Scholar]
- Li, P.; Li, Z.; Li, D.; Li, J. Report of Leaf Spot on Pueraria lobata Caused by Bipolaris sorokiniana in China. Plant Dis. 2019, 103, 1040. [Google Scholar] [CrossRef]
- Yan, M.F.; Zou, M.F.; Liu, B.; Wang, Y.X.; Xiong, G.H.; Jiang, J.X. First report of leaf spot caused by Pseudocercospora puerariicola on kudzu (Pueraria lobata) in Jiangxi China. J. Plant Pathol. 2018, 100, 609. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, I.; Kissen, R.; Bones, A.M. Phytoalexins in defense against pathogens. Trends Plant Sci. 2012, 17, 73–90. [Google Scholar] [CrossRef]
- Cui, T.H.; Tang, S.Y.; Liu, C.B.; Li, Z.J.; Zhu, Q.; You, J.H.; Si, X.X.; Zhang, F.M.; He, P.; Liu, Z.H.; et al. Three new isoflavones from the Pueraria montana var. lobata (Willd.) and their bioactivities. Nat. Prod. Res. 2018, 32, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Suntichaikamolkul, N.; Tantisuwanichkul, K.; Prombutara, P.; Kobtrakul, K.; Zumsteg, J.; Wannachart, S.; Schaller, H.; Yamazaki, M.; Saito, K.; De-Eknamkul, W.; et al. Transcriptome analysis of Pueraria candollei var. mirifica for gene discovery in the biosyntheses of isoflavones and miroestrol. BMC Plant Biol. 2019, 19, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, C.F.; Zhou, Z.L.; Zhang, Y.S. Identification of Three (Iso)flavonoid Glucosyltransferases from Pueraria lobata. Front. Plant Sci. 2019, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.Y.; Yuan, G.Q.; He, L.F.; Xiao, D.; Wang, A.Q. Investigation on pseudorust disease of guige series varieties. Guangdong Agric. Sci. 2019, 12, 83–88. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, X.D.; Guo, L.J.; Zeng, D.Q.; He, L.F.; Xiao, D.; Wang, A.Q. Study on quality analysis of pueraria germplasm resources by membership function method. J. Chin. Med. Mater. 2017, 40, 1314–1317. [Google Scholar] [CrossRef]
- Eva, M.; Ruben, V.; Wout, B.; Antonio, M. The role of the secondary cell wall in plant resistance to pathogens. Front. Plant Sci. 2014, 5, 358. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.Y.; Liu, Z.H.; Zhu, L.; Zhang, C.J.; Chen, Y.; Zhou, Y.; Li, F.G.; Li, X.B. Overexpression of cotton (Gossypium hirsutum) dirigent1 gene enhances lignification that blocks the spread of Verticillium dahliae. Acta Biochim. Biophys. Sin. 2012, 44, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Gimenez-Ibanez, S.; Boter, M.; Fernández-Barbero, G.; Chini, A.; Rathjen, J.P.; Solano, R. The bacterial effector HopX1 targets JAZ transcriptional repressors to activate jasmonate signaling and promote infection in Arabidopsis. PLoS Biol. 2014, 12, e1001792. [Google Scholar] [CrossRef] [Green Version]
- Xin, X.F.; Sheng, Y.H. Pseudomonas syringae pv. tomato DC3000: A Model Pathogen for Probing Disease Susceptibility and Hormone Signaling in Plants. Annu. Rev. Phytopathol. 2013, 51, 473–498. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, Y.Y.; Du, M.; Zhang, X.; Guo, Y.; Li, C.; Zhou, J.M. An Arabidopsis plasma membrane proton ATPase modulates JA signaling and is exploited by the pseudomonas syringae effector protein AvrB for stomatal invasion. Plant Cell 2015, 27, 2032–2041. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.C.; Liang, F.; Zhang, J.R.; Wang, F.; Liu, D.Q.; Wang, H.Y. Transgenic expression of TaTLP1, a thaumatin-like protein gene, reduces susceptibility to common root rot and leaf rust in wheat. Crop J. 2021, 9, 1214–1218. [Google Scholar] [CrossRef]
- Dolatabadian, A.; Bayer, P.E.; Tirnaz, S.; Hurgobin, B.; Edwards, D.; Batley, J. Characterization of disease resistance genes in the Brassica napus pangenome reveals significant structural variation. Plant Biotechnol. J. 2020, 18, 969–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.X.; Zhong, Y.; Huang, K.H.; Wang, S.H.; Cheng, Q.K.; Cheng, Z.M. Differential expression of 12 NBS-encoding genes in two apple cultivars in response to Alternaria alternata f. sp. mali infection. Can. J. Plant Sci. 2018, 98, 279–287. [Google Scholar] [CrossRef]
- Cantu, D.; Vicente, A.R.; Labavitch, J.M.; Bennett, A.B.; Powell, A.L.T. Strangers in the matrix: Plant cell walls and pathogen susceptibility. Trends Plant Sci. 2008, 13, 610–617. [Google Scholar] [CrossRef]
- Hückelhoven, R. Cell Wall-Associated Mechanisms of Disease Resistance and Susceptibility. Annu. Rev. Phytopathol. 2007, 45, 101–127. [Google Scholar] [CrossRef] [PubMed]
- Underwood, W.; Somerville, S.C. Focal accumulation of defences at sites of fungal pathogen attack. J. Exp. Bot. 2008, 59, 3501–3508. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.M.; Panstruga, R. Pathogenomics of fungal plant parasites: What have we learnt about pathogenesis? Curr. Opin. Plant Biol. 2011, 14, 392–399. [Google Scholar] [CrossRef]
- Ramírez, V.; Agorio, A.; Coego, A.; García-Andrade, J.; Hernández, M.J.; Balaguer, B.; Ouwerkerk, P.B.F.; Zarra, I.; Vera, P. MYB46 modulates disease susceptibility to Botrytis cinerea in Arabidopsis. Plant Physiol. 2011, 155, 1920–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camilo, H.B.; Feng, D.X.; Hu, J.; Andrea, S.V.; Deslandes, L.; Francisco, L.; Marta, B.L.; Keller, H.; Barlet, X.; Clara, S.R.; et al. Impairment of cellulose synthases required for Arabidopsis secondary cell wall formation enhances disease resistance. Plant Cell 2007, 19, 890–903. [Google Scholar] [CrossRef] [Green Version]
- Yuzuki, M.; Nafisi, M.; Verhertbruggen, Y.; Orfila, C.; Gille, S.; Rautengarten, C.; Cherk, C.; Marcus, S.E.; Somerville, S.; Pauly, M.; et al. Loss-of-function mutation of REDUCED WALL ACETYLATION2 in Arabidopsis leads to reduced cell wall acetylation and increased resistance to Botrytis cinerea. Plant Physiol. 2011, 155, 1068–1078. [Google Scholar] [CrossRef] [Green Version]
- Møller, I.M.; Jensen, P.E.; Hansson, A. Oxidative Modifications to Cellular Components in Plants. Annu. Rev. Plant Biol. 2007, 58, 459–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, R.G.; Jonathan, D.G.J. Hormone (Dis)harmony Moulds Plant Health and Disease. Science 2009, 324, 750–752. [Google Scholar] [CrossRef]
- Bostock, R.M. Signal crosstalk and induced resistance: Straddling the line between cost and beneft. Annu. Rev. Phytopathol. 2005, 43, 545–580. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.J.; Guo, X.; Zhao, D.; Zhang, Q.; Jiang, Y.Z.; Wang, Y.T.; Peng, X.; Wei, Y.; Zhai, Z.F.; Zhao, W.; et al. Cloning and expression profling of the PacSnRK2 and PacPP2C gene families during fruit development, ABA treatment, and dehydration stress in sweet cherry. Plant Physiol. Biochem. 2017, 119, 275–285. [Google Scholar] [CrossRef]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef]
- Chini, A.; Fonseca, S.; Chico, J.M.; Fernández-Calvo, P.; Solano, R. The ZIM domain mediates homo- and heteromeric interactions between arabidopsis JAZ protein. Plant J. 2009, 59, 77–87. [Google Scholar] [CrossRef]
- Staswick, P.E. JAZing up jasmonate signaling. Trends Plant Sci. 2007, 13, 66–71. [Google Scholar] [CrossRef]
- Lorenzo, O.; Priquerasr, R.; Shez-serrano, J.J.; Solano, R. Ethylene response factorl intergrates signals from ethylene and jasmonate pathways in plant defense. Plant Cell 2003, 15, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Díaz, J.; Arjen, T.H.; van Kan Jan, A.L. The role of ethylene and wound signaling in resistance of tomato to Botrytis cinerea. Plant Physiol. 2002, 129, 1341–1351. [Google Scholar] [CrossRef] [Green Version]
- Helliwell, E.E.; Wang, Q.; Yang, Y.N. Ethylene Biosynthesis and Signaling Is Required for Rice Immune Response and Basal Resistance Against Magnaporthe oryzae Infection. Mol. Plant-Microbe Interact. MPMI 2016, 29, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Long, Q.; Xie, Y.; He, Y.R.; Li, Q.; Zou, X.P.; Chen, S.C. Abscisic Acid Promotes Jasmonic Acid Accumulation and Plays a Key Role in Citrus Canker Development. Front. Plant Sci. 2019, 10, 1634. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Zhang, L.D.; Chen, J.B. Physiological analysis and transcriptome comparison of two muskmelon (Cucumis melo L.) cultivars in response to salt stress. Genet. Mol. Res. 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Zeng, H.Q.; Xu, L.Q.; Singh, A.; Wang, H.Z.; Du, L.Q.; Poovaiah, B.W. Involvement of calmodulin and calmodulin-like proteins in plant responses to abiotic stresses. Front. Plant Sci. 2015, 6, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcec, M.J.; Gilroy, S.; Poovaiah, B.W.; Tanaka, K. Mutual interplay of Ca2+ and ROS signaling in plant immune response. Plant Sci. 2019, 283, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.; Guo, J.; Liu, P.; He, F.X.; Wan, C.P.; Islam, M.A.; Tyler, B.M.; Kang, Z.S.; Guo, J. Stripe Rust Effector Pst GSRE1 Disrupts Nuclear Localization of ROS-Promoting Transcription Factor Ta LOL2 to Defeat ROS-Induced Defense in Wheat. Mol. Plant 2019, 12, 1624–1638. [Google Scholar] [CrossRef]
- Xu, Q.; Tang, C.L.; Wang, X.D.; Sun, S.T.; Zhao, J.R.; Kang, Z.S.; Wang, X.J. An effector protein of the wheat Stripe rust fungus targets chloroplasts and suppresses chloroplast function. Nat. Commun. 2019, 10, 5571. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Q.; Chen, T.T.; Yin, X.; Xiang, G.Q.; Peng, J.; Fu, Q.Q.; Li, M.Y.; Shang, B.X.; Ma, H.; Liu, G.T.; et al. A Plasmopara viticola RXLR effector targets a chloroplast protein Psb P to inhibit ROS production in grapevine. Plant J. 2021, 106, 1557–1570. [Google Scholar] [CrossRef]
- Peter, L.D.H.; Laurence, M.B.; Ann, M. Plant lectins: The ties that bind in root symbiosis and plant defense. Mol. Genet. Genom. 2009, 282, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.S.; Hwang, B.K. The pepper mannose- binding lectin gene CaMBL1 is required to regulate cell death and defense responses to microbial pathogens. Plant Physiol. 2011, 155, 447–463. [Google Scholar] [CrossRef] [Green Version]
- Bouwmeester, K.; De Sain, M.; Weide, R.; Gouget, A.; Klamer, S.; Canut, H.; Govers, F. The lectin receptor kinase LecRK-I. 9 is a novel Phytophthora resistance component and a potential host target for a RXLR effector. PLoS Pathog. 2011, 7, e1001327–e1001397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seunghwan, K.; Eric, J.W.; Jihwan, P.; Sik, K.M.; Gu, L.J.; Woo, K.L. Combined Analysis of the Time-Resolved Transcriptome and Proteome of Plant Pathogen Xanthomonas oryzae pv. oryzae. Front. Microbiol. 2021, 12, 664857. [Google Scholar] [CrossRef]
- Han, S.W.; Lee, M.A.; Yoo, Y.; Cho, M.H.; Lee, S.W. Genome-wide Screening to Identify Responsive Regulators Involved in the Virulence of Xanthomonas oryzae pv. oryzae. Plant Pathol. J. 2019, 35, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Min, C.W.; Son, S.M.; Lee, G.H.; Jang, J.W.; Kwon, S.W.; Park, S.R.; Kim, S.T. Comparative proteome profiling of susceptible and resistant rice cultivars identified an arginase involved in rice defense against Xanthomonas oryzae pv. oryzae. Plant Physiol. Biochem. 2021, 171, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Number | Ratio% |
|---|---|---|
| GO | 32,304 | 45.57 |
| KEGG | 25,504 | 35.97 |
| Pfam | 30,022 | 42.35 |
| swissprot | 26,129 | 36.86 |
| eggNOG | 39,537 | 55.77 |
| KOG | 25,504 | 35.97 |
| NR | 39,828 | 56.18 |
| Intersection | 7445 | 10.50 |
| Overall | 59,701 | 84.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Huang, X.; Guo, L.; He, L.; Xiao, D.; Zhan, J.; Wang, A.; Liang, R. Comparative Transcriptome Analysis Provides Insights into the Resistance in Pueraria [Pueraria lobata (Willd.) Ohwi] in Response to Pseudo-Rust Disease. Int. J. Mol. Sci. 2022, 23, 5223. https://doi.org/10.3390/ijms23095223
Huang X, Huang X, Guo L, He L, Xiao D, Zhan J, Wang A, Liang R. Comparative Transcriptome Analysis Provides Insights into the Resistance in Pueraria [Pueraria lobata (Willd.) Ohwi] in Response to Pseudo-Rust Disease. International Journal of Molecular Sciences. 2022; 23(9):5223. https://doi.org/10.3390/ijms23095223
Chicago/Turabian StyleHuang, Xinlu, Xiaoxi Huang, Lijun Guo, Longfei He, Dong Xiao, Jie Zhan, Aiqin Wang, and Renfan Liang. 2022. "Comparative Transcriptome Analysis Provides Insights into the Resistance in Pueraria [Pueraria lobata (Willd.) Ohwi] in Response to Pseudo-Rust Disease" International Journal of Molecular Sciences 23, no. 9: 5223. https://doi.org/10.3390/ijms23095223
APA StyleHuang, X., Huang, X., Guo, L., He, L., Xiao, D., Zhan, J., Wang, A., & Liang, R. (2022). Comparative Transcriptome Analysis Provides Insights into the Resistance in Pueraria [Pueraria lobata (Willd.) Ohwi] in Response to Pseudo-Rust Disease. International Journal of Molecular Sciences, 23(9), 5223. https://doi.org/10.3390/ijms23095223