
Overexpression of VEGF in the MOPC 315 Plasmacytoma Induces Tumor Immunity in Mice

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
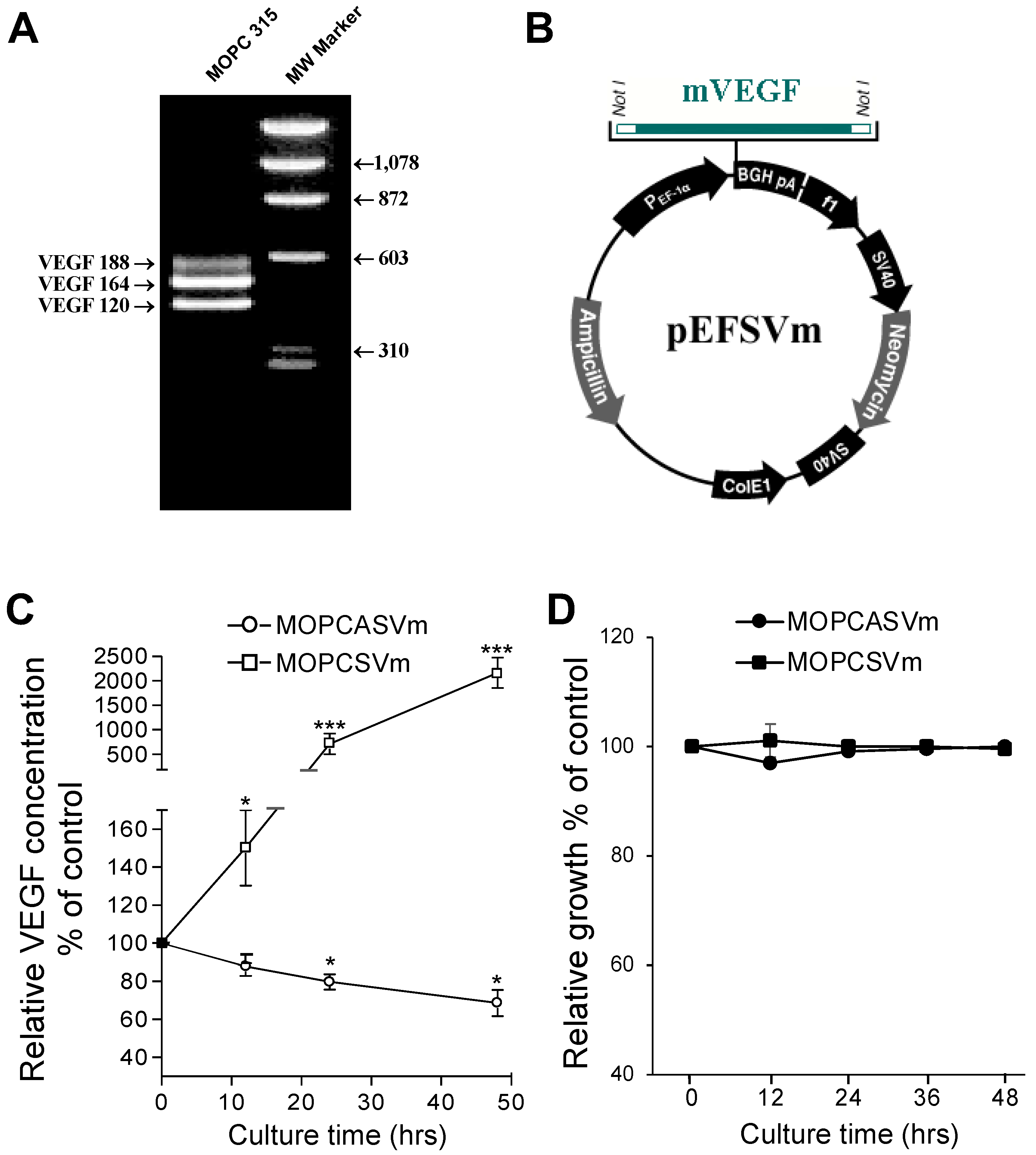
2.1. Ectopic Expression of VEGF Had No Effect on Plasmacytoma Cell Growth In Vitro
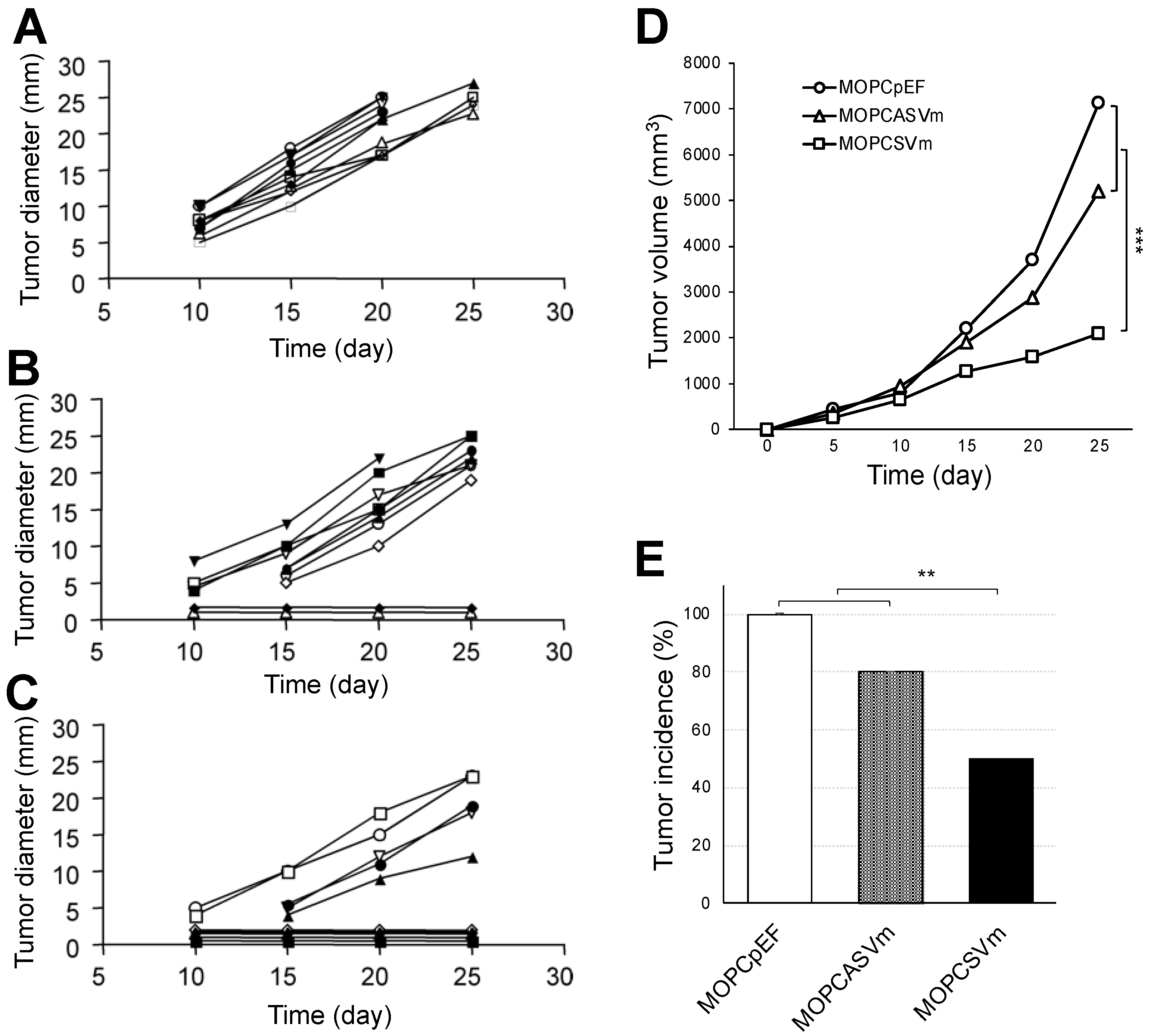
2.2. Reduced Tumorigenicity of VEGF-Expressing MOPC 315 Transfectants in BALB/c Mice
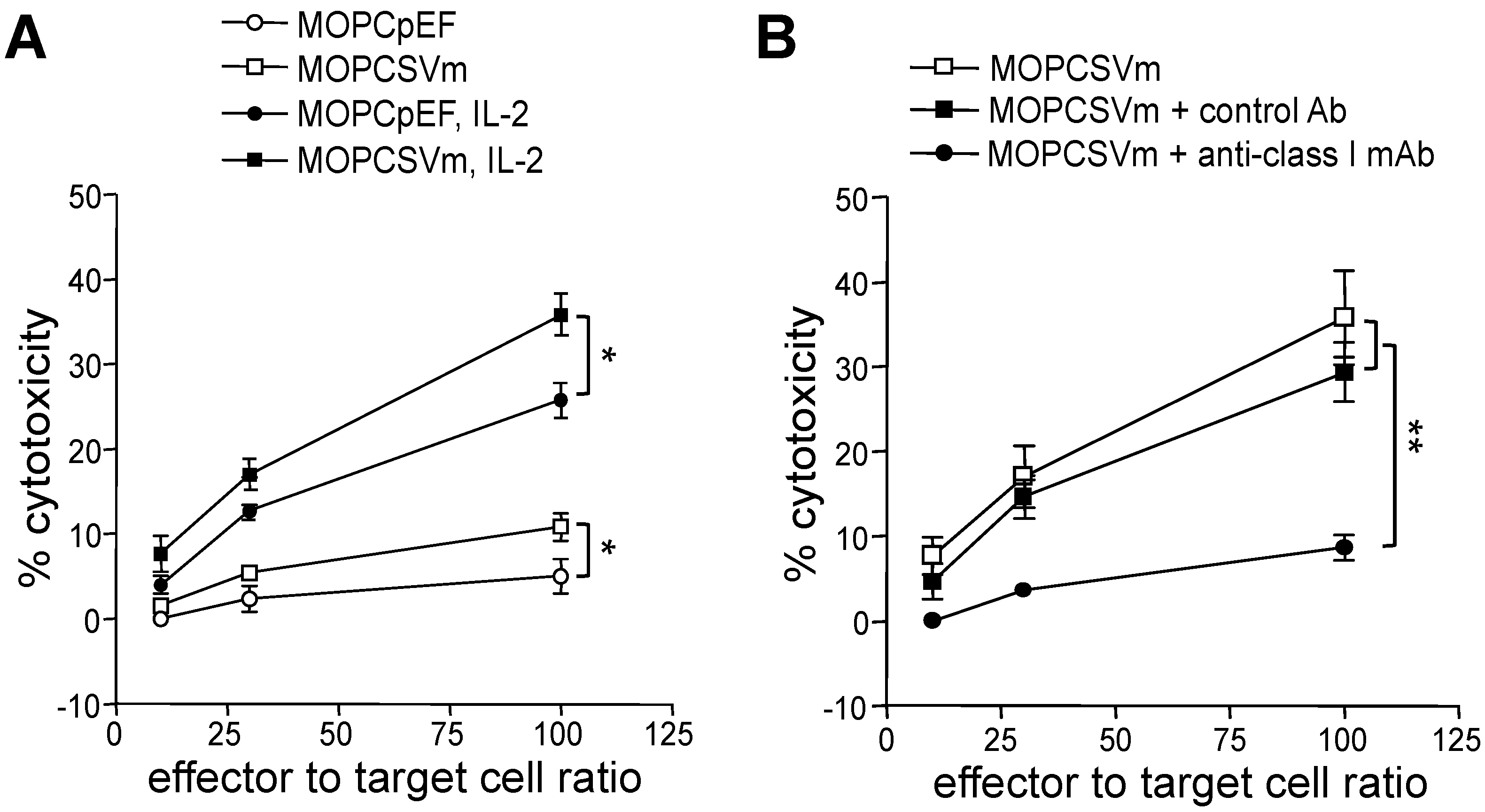
2.3. Development of Cell-Mediated Immunity in Mice Implanted with MOPCSVm Cells
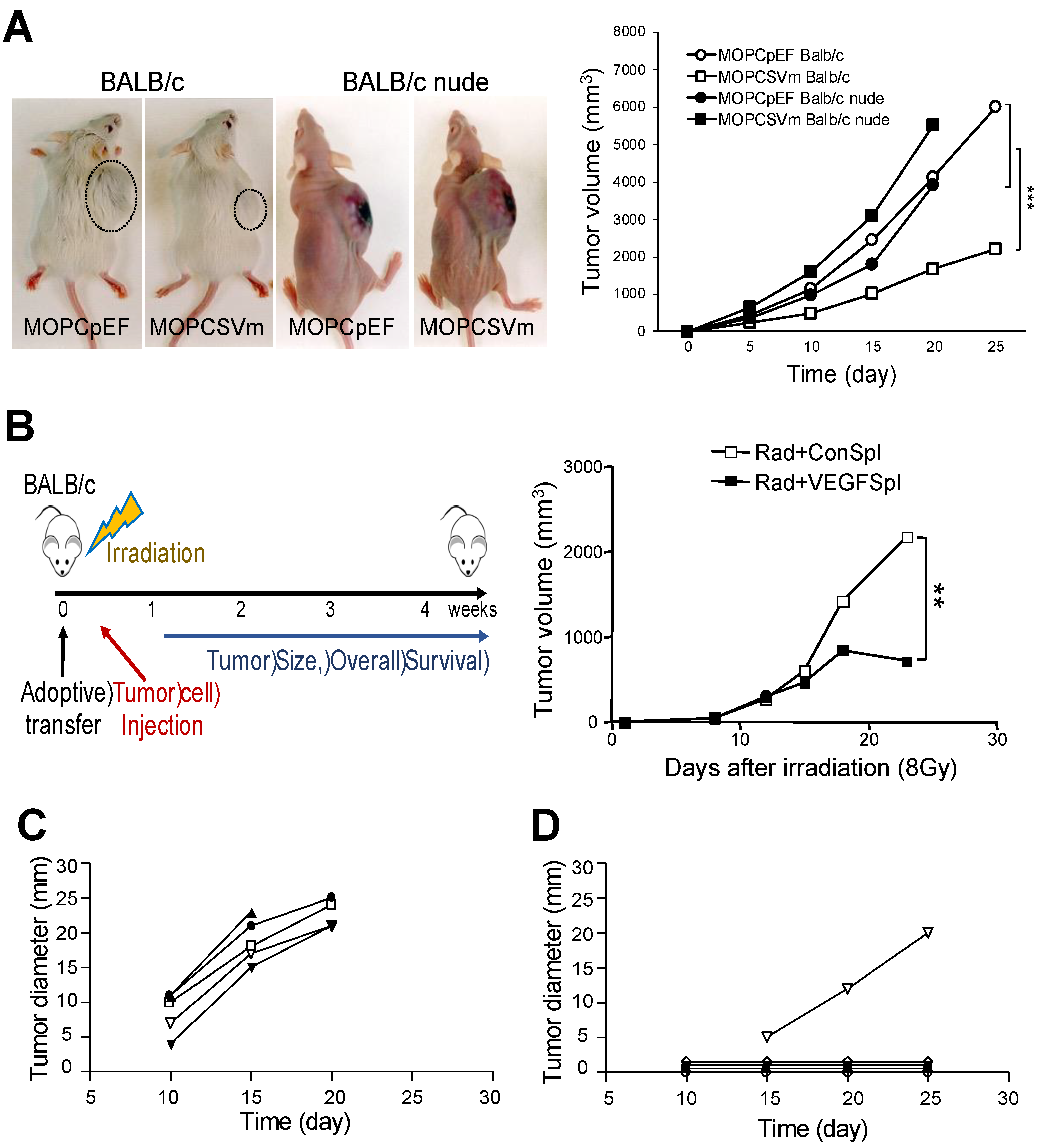
2.4. Regression of MOPCSVm Tumors Was T-Cell-Dependent
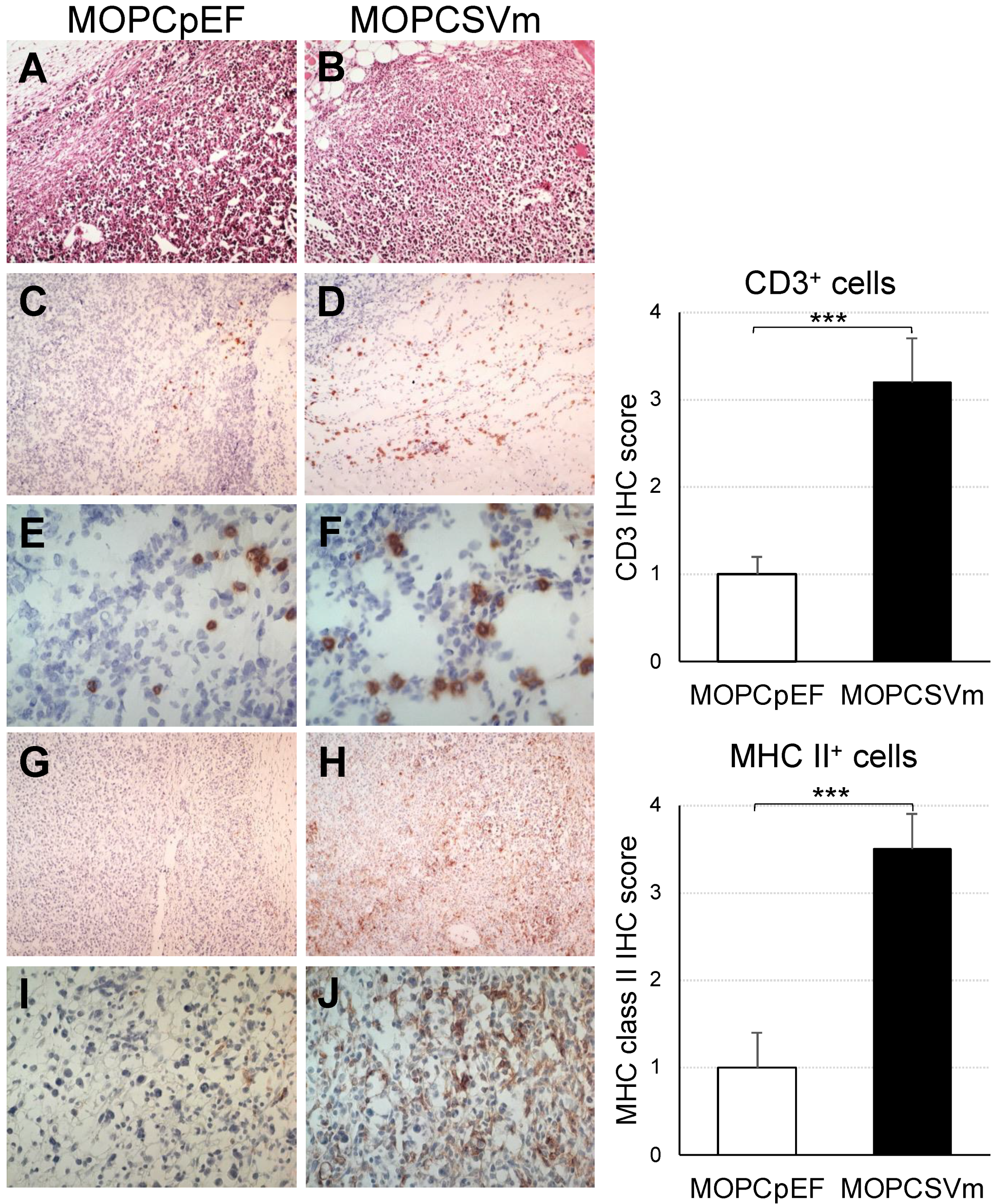
2.5. Diffuse Infiltration of Leukocytes around MOPCSVm
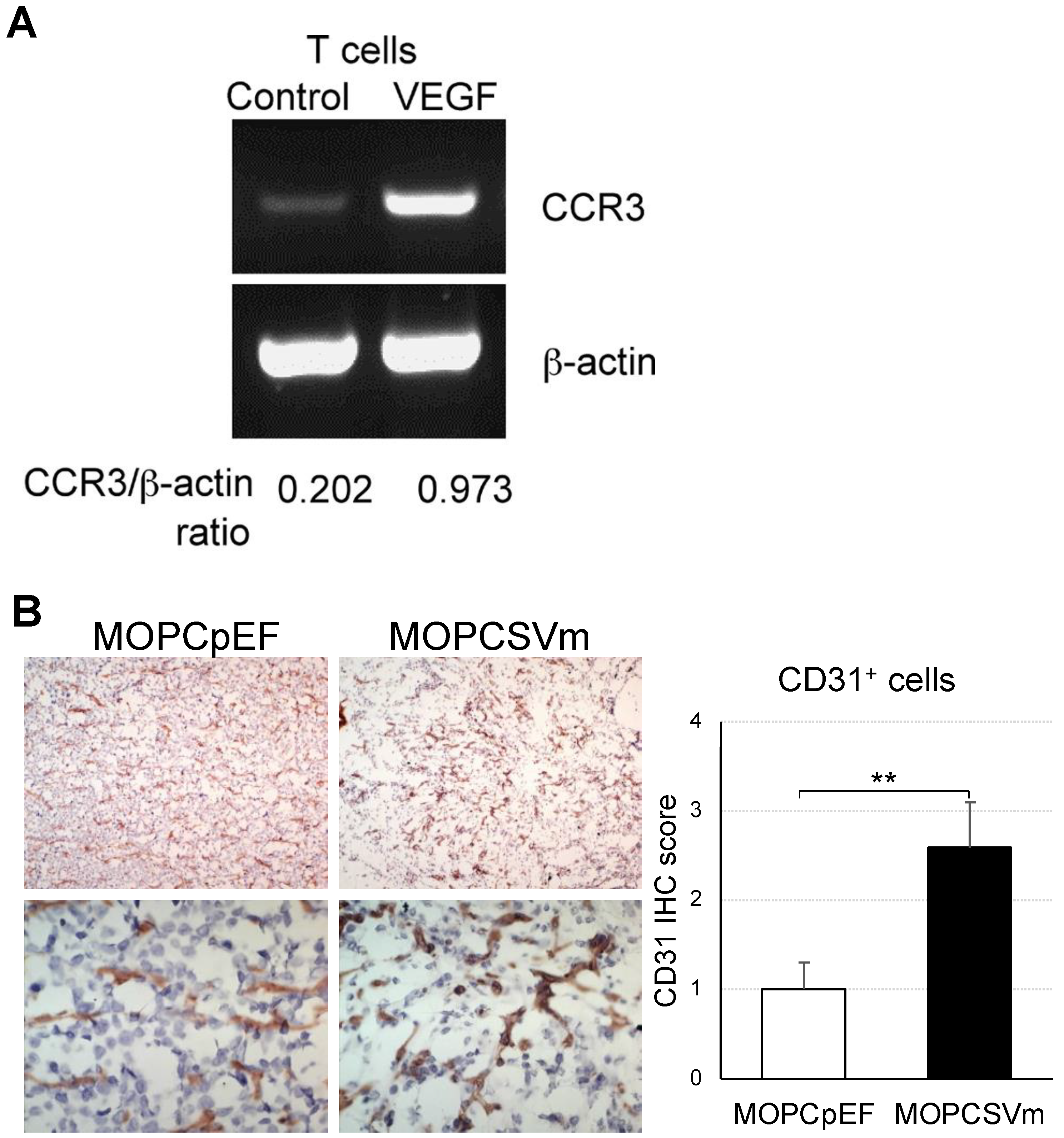
2.6. Induction of Chemokine Receptor Expression and Angiogenesis by VEGF
3. Discussion
4. Materials and Methods
4.1. Tumor Cell Lines and Cell Culture Conditions
4.2. RNA Isolation and RT-PCR
4.3. Construction of VEGF Vector
4.4. Transfection and Quantification of VEGF
4.5. Assay of In Vitro Growth Rate
4.6. Tumorigenicity and Immunization of Animals
4.7. Mixed Lymphocyte Tumor Cell Cultures (MLTC)
4.8. Cytotoxicity Assay
4.9. Neutralization Test
4.10. Adoptive Transfer
4.11. Immunohistochemical Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Millauer, B.; Wizigmann-Voos, S.; Schnurch, H.; Martinez, R.; Moller, N.P.; Risau, W.; Ullrich, A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72, 835–846. [Google Scholar] [CrossRef]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.C.; Abraham, J.A. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar] [CrossRef]
- Park, J.E.; Keller, G.A.; Ferrara, N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, D.T.; Kuroki, M.; Deutsch, U.; Ng, Y.S.; Adamis, A.P.; D’Amore, P.A. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J. Biol. Chem. 1996, 271, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Jurisic, V. Multiomic analysis of cytokines in immuno-oncology. Expert. Rev. Proteom. 2020, 17, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.J.; Wang, J.; Deng, X.Y.; Xiong, F.; Zhang, S.S.; Gong, Z.J.; Li, X.Y.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Canc. Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, W.T.; Richter, L.; Frutiger, Y.; Grogan, T.M. E 426 xpression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999, 59, 728–733. [Google Scholar]
- Dankbar, B.; Padro, T.; Leo, R.; Feldmann, B.; Kropff, M.; Mesters, R.M.; Serve, H.; Berdel, W.E.; Kienast, J. Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood 2000, 95, 2630–2636. [Google Scholar] [CrossRef]
- Aguayo, A.; Kantarjian, H.; Manshouri, T.; Gidel, C.; Estey, E.; Thomas, D.; Koller, C.; Estrov, Z.; O’Brien, S.; Keating, M.; et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood 2000, 96, 2240–2245. [Google Scholar] [CrossRef]
- Mesa, R.A.; Hanson, C.A.; Rajkumar, S.V.; Schroeder, G.; Tefferi, A. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood 2000, 96, 3374–3380. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Fonseca, R.; Greipp, P.R.; Rajkumar, S.V. Expression of VEGF and its receptors by myeloma cells. Leukemia 2003, 17, 2025–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF Drugs in the Treatment of Multiple Myeloma Patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Leong, T.; Roche, P.C.; Fonseca, R.; Dispenzieri, A.; Lacy, M.Q.; Lust, J.A.; Witzig, T.E.; Kyle, R.A.; Gertz, M.A.; et al. Prognostic value of bone marrow angiogenesis in multiple myeloma. Clin. Cancer Res. 2000, 6, 3111–3116. [Google Scholar] [PubMed]
- Paydas, S.; Zorludemir, S.; Baslamisli, F.; Tuncer, I. Vascular endothelial growth factor (VEGF) expression in plasmacytoma. Leuk. Lymphoma 2002, 43, 139–143. [Google Scholar] [CrossRef]
- Swelam, W.M.; Al Tamimi, D.M. Biological impact of vascular endothelial growth factor on vessel density and survival in multiple myeloma and plasmacytoma. Pathol. Res. Pract. 2010, 206, 753–759. [Google Scholar] [CrossRef]
- Steinman, R.M. Dendritic cells in vivo: A key target for a new vaccine science. Immunity 2008, 29, 319–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M. Decisions about Dendritic Cells: 470 Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B.R. Targeting veGF/veGFR to Modulate Antitumor immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Long, J.H.; Hu, Z.Q.; Xue, H.; Wang, Y.; Chen, J.; Tang, F.Z.; Zhou, J.; Liu, L.N.; Qiu, W.; Zhang, S.C.; et al. Vascular endothelial growth factor (VEGF) impairs the motility and immune function of human mature dendritic cells through the VEGF receptor 2-RhoA-cofilin1 pathway. Cancer Sci. 2019, 110, 2357–2367. [Google Scholar] [CrossRef]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, M.J.; Buendia, P.; Velasco, F.; Aguirre, M.A.; Barbarroja, N.; Torres, L.A.; Khamashta, M.; Lopez-Pedrera, C. Vascular endothelial growth factor expression in monocytes from patients with primary antiphospholipid syndrome. J. Thromb. Haemost. 2006, 4, 2461–2469. [Google Scholar] [CrossRef]
- Hidman, J.; Larsson, A.; Thulin, M.; Karlsson, T. Increased Plasma GDF15 Is Associated with Altered Levels of Soluble VEGF Receptors 1 and 2 in Symptomatic Multiple Myeloma. Acta Haematol. 2021, 1–8. [Google Scholar] [CrossRef]
- Freeman, M.R.; Schneck, F.X.; Gagnon, M.L.; Corless, C.; Soker, S.; Niknejad, K.; Peoples, G.E.; Klagsbrun, M. Peripheral blood T lymphocytes and lymphocytes infiltrating human cancers express vascular endothelial growth factor: A potential role for T cells in angiogenesis. Cancer Res. 1995, 55, 4140–4145. [Google Scholar]
- Iijima, K.; Yoshikawa, N.; Nakamura, H. Activation-induced expression of vascular permeability factor by human peripheral T cells: A non-radioisotopic semiquantitative reverse transcription-polymerase chain reaction assay. J. Immunol. Methods 1996, 196, 199–209. [Google Scholar] [CrossRef]
- Matsuyama, W.; Kubota, R.; Hashiguchi, T.; Momi, H.; Kawabata, M.; Nakagawa, M.; Arimura, K.; Osame, M. Purified protein derivative of tuberculin upregulates the expression of vascular endothelial growth factor in T lymphocytes in vitro. Immunology 2002, 106, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; Gabrilovich, D.I.; Sempowski, G.D.; Kisseleva, E.; Parman, K.S.; Nadaf, S.; Carbone, D.P. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 2003, 101, 4878–4886. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8(+) T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Mor, F.; Quintana, F.J.; Cohen, I.R. Angiogenesis-inflammation cross-talk: Vascular endothelial factor is secreted by activated T cells and induces Th1 polarization. J. Immunol. 2004, 172, 4618–4623. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Hoerning, A.; Datta, D.; Edelbauer, M.; Stack, M.P.; Calzadilla, K.; Pal, S.; Briscoe, D.M. Cutting Edge: Vascular Endothelial Growth Factor-Mediated Signaling in Human CD45RO(+) CD4(+) T Cells Promotes Akt and ERK Activation and Costimulates IFN534 gamma Production. J. Immunol. 2010, 184, 545–549. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.L.; Iragavarapu-Charyulu, V.; Gunja-Smith, Z.; Herbert, L.M.; Grosso, J.F.; Lopez, D.M. Up538 regulation of matrix metalloproteinase-9 in T lymphocytes of mammary tumor bearers: Role of vascular endothelial growth factor. J. Immunol. 2003, 171, 4340–4351. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Velica, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1 alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Yamamoto, L.; Amodio, N.; Gulla, A.; Anderson, K.C. Harnessing the Immune System Against Multiple Myeloma: Challenges and Opportunities. Front. Oncol. 2020, 10, 606368. [Google Scholar] [CrossRef]
- Colombo, M.; Giannandrea, D.; Lesma, E.; Basile, A.; Chiaramonte, R. Extracellular Vesicles Enhance Multiple Myeloma Metastatic Dissemination. Int. J. Mol. Sci. 2019, 20, 3236. [Google Scholar] [CrossRef] [Green Version]
- Konjevic, G.; Vuletic, A.; Mirjacic Martinovic, K.; Colovic, N.; Colovic, M.; Jurisic, V. Decreased CD161 activating and increased CD158a inhibitory receptor expression on NK cells underlies impaired NK cell cytotoxicity in patients with multiple myeloma. J. Clin. Pathol. 2016, 69, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Venglar, O.; Bago, J.R.; Motais, B.; Hajek, R.; Jelinek, T. Natural Killer Cells in the Malignant Niche of Multiple Myeloma. Front. Immunol. 2021, 12, 816499. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.Q.; Zhou, J.; Sasada, T.; Zeng, W.Y.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.Q.; Giobbie-Hurder, A.; Liao, X.Y.; Lawrence, D.; McDermott, D.; Zhou, J.; Rodig, S.; Hodi, F.S. VEGF Neutralization Plus CTLA-4 Blockade Alters Soluble and Cellular Factors Associated with Enhancing Lymphocyte Infiltration a 561 and Humoral Recognition in Melanoma. Cancer Immunol. Res. 2016, 4, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Satooka, H.; Ishigaki, H.; Todo, K.; Terada, K.; Agata, Y.; Itoh, Y.; Ogasawara, K.; Hirata, T. Characterization of tumour-infiltrating lymphocytes in a tumour rejection cynomolgus macaque model. Sci. Rep. 2020, 10, 8414. [Google Scholar] [CrossRef]
- Danilova, E.; Skrindo, I.; Gran, E.; Hales, B.J.; Smith, W.A.; Jahnsen, J.; Johansen, F.E.; Jahnsen, F.L.; Baekkevold, E.S. A role for CCL28-CCR3 in T-cell homing to the human upper airway mucosa. Mucosal. Immunol. 2015, 8, 107–114. [Google Scholar] [CrossRef]
- Claffey, K.P.; Brown, L.F.; del Aguila, L.F.; Tognazzi, K.; Yeo, K.T.; Manseau, E.J.; Dvorak, H.F. Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis, and experimental metastasis. Cancer Res. 1996, 56, 172–181. [Google Scholar]
- Jurisic, V.; Srdic, T.; Konjevic, G.; Markovic, O.; Colovic, M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med. Oncol. 2007, 24, 312–317. [Google Scholar] [CrossRef]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1alpha in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8, 1597. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Park, S.; Min, W.S.; Kim, H.J. Restoration of natural killer cell cytotoxicity by VEGFR-3 inhibition in myelogenous leukemia. Cancer Lett. 2014, 354, 281–289. [Google Scholar] [CrossRef]
- Yousefi, Y.; Mortezania, Z.; Abolmolouki, M.; Daemi, A. A novel form of anti-angiogenic molecular antibody drug induces apoptosis in myeloma cells after cultivation upon endothelial feeder cells. Leuk. Res. 2021, 108, 106617. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Joo, H.G.; Chung, I.S.; Chung, H.Y.; Woo, H.J.; Yun, Y.S. Inhibition of interleukin-10 (IL-10) production from MOPC 315 tumor cells by IL-10 antisense oligodeoxynucleotides enhances cell-mediated immune responses. Cancer Immunol. Immunother. 2000, 49, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.; Beems, M.V.; Tatar, A.J.; Sen, S.; Srinand, P.; Suresh, M.; Luther, T.K.; Cho, C.S. Co-transfer of tumor-specific effector and memory CD8+ T cells enhances the efficacy of adoptive melanoma immunotherapy in a mouse model. J. Immunother. Cancer 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Kim, M.; Kim, H.S.; Kang, H.J.; Kim, H.J.; Min, S.K.; Lee, Y.K. CD3 and CD20 Immunohistochemical Staining Patterns of Bone Marrow-Infiltrating Malignant Lymphoma Cells. Ann. Clin. Lab. Sci. 2017, 47, 136–143. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-G.; Choi, S.H.; Letterio, J.J.; Song, J.-Y.; Huang, A.Y. Overexpression of VEGF in the MOPC 315 Plasmacytoma Induces Tumor Immunity in Mice. Int. J. Mol. Sci. 2022, 23, 5235. https://doi.org/10.3390/ijms23095235
Kim B-G, Choi SH, Letterio JJ, Song J-Y, Huang AY. Overexpression of VEGF in the MOPC 315 Plasmacytoma Induces Tumor Immunity in Mice. International Journal of Molecular Sciences. 2022; 23(9):5235. https://doi.org/10.3390/ijms23095235
Chicago/Turabian StyleKim, Byung-Gyu, Sung Hee Choi, John J. Letterio, Jie-Young Song, and Alex Y. Huang. 2022. "Overexpression of VEGF in the MOPC 315 Plasmacytoma Induces Tumor Immunity in Mice" International Journal of Molecular Sciences 23, no. 9: 5235. https://doi.org/10.3390/ijms23095235
APA StyleKim, B.-G., Choi, S. H., Letterio, J. J., Song, J.-Y., & Huang, A. Y. (2022). Overexpression of VEGF in the MOPC 315 Plasmacytoma Induces Tumor Immunity in Mice. International Journal of Molecular Sciences, 23(9), 5235. https://doi.org/10.3390/ijms23095235