
The Adaptability of Chromosomal Instability in Cancer Therapy and Resistance


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
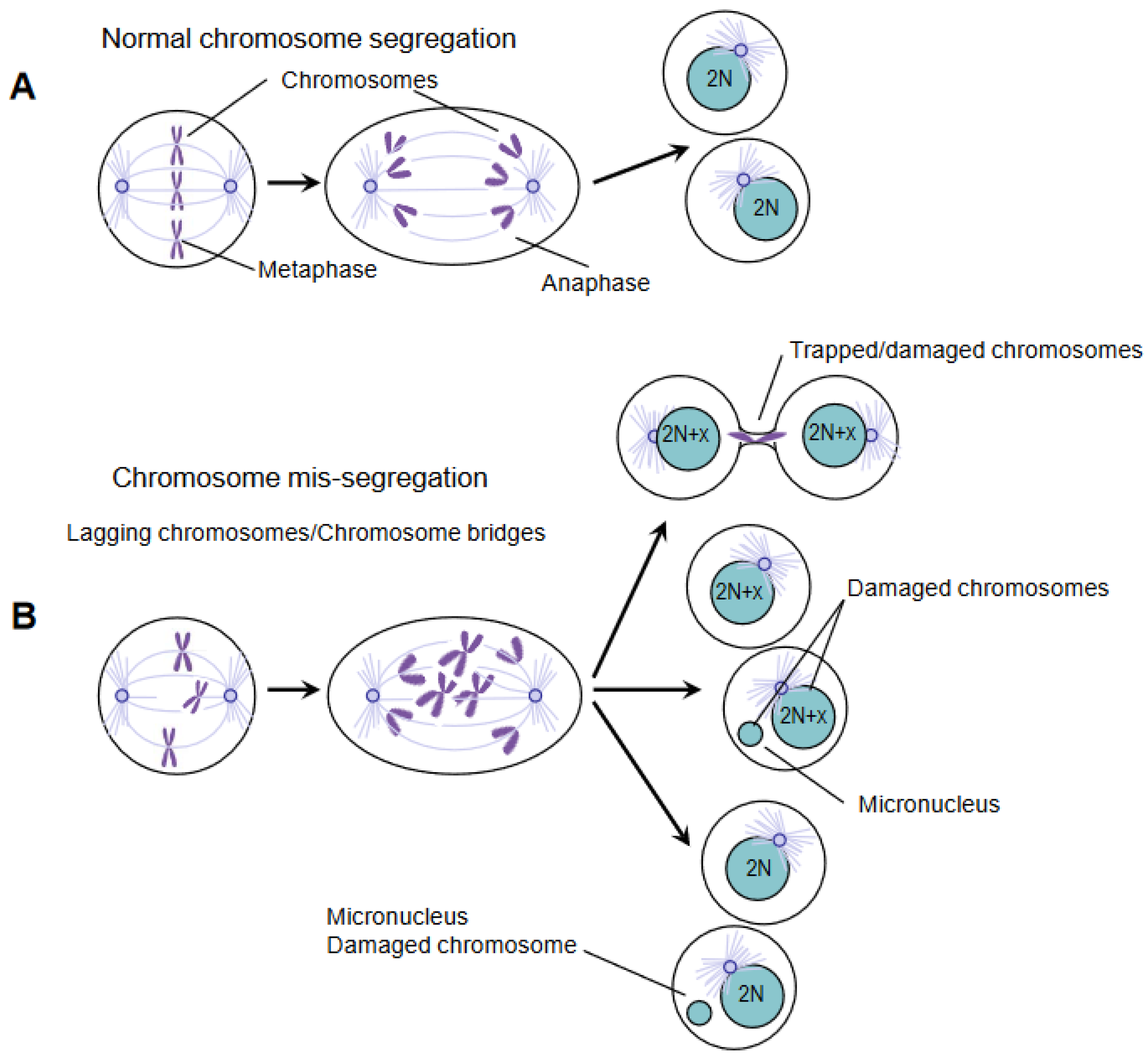
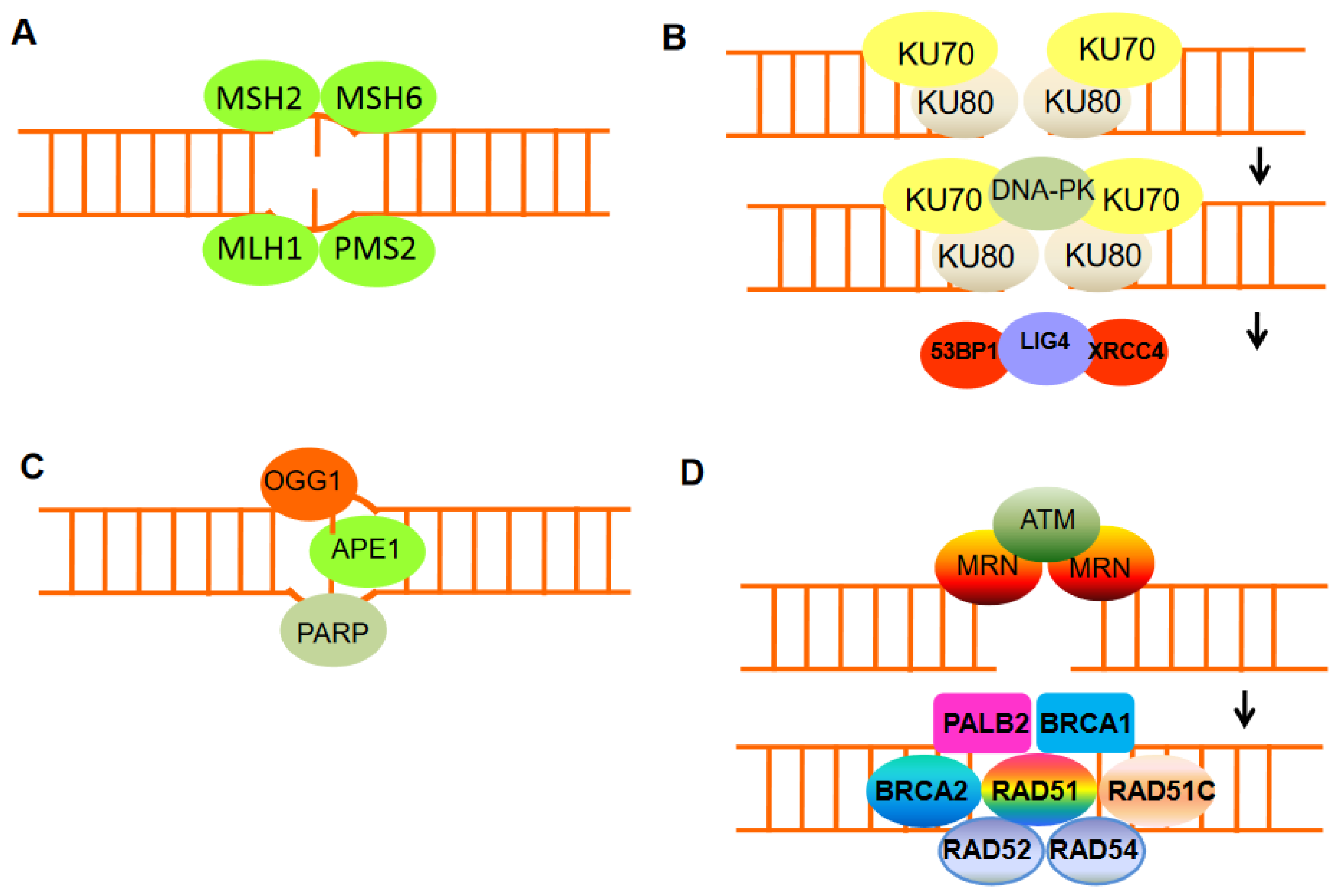
2. DNA Damage and Chromosomal Instability
3. DDR Components Are Targets of Chemotherapy Drugs
4. Importance of DDR and Chromatin Remodeling in Therapeutics Response and Resistance
5. DNA Damage and Immunotherapy
6. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The DNA damage response during DNA replication. Curr. Opin. Cell Biol. 2005, 17, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. BRCA1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.; Dürrbaum, M.; Kuffer, C.; Müller, S.; Zaman, C.J.R.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [Green Version]
- Rowald, K.; Mantovan, M.; Passos, J.; Buccitelli, C.; Mardin, B.R.; Korbel, J.O.; Jechlinger, M.; Sotillo, R. Negative selection and chromosome instability induced by Mad2 overexpression delay breast cancer but facilitate oncogene-independent outgrowth. Cell Rep. 2016, 15, 2679–2691. [Google Scholar] [CrossRef] [Green Version]
- Sotillo, R.; Schvartzman, J.-M.; Socci, N.D.; Benezra, R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 2010, 464, 436–440. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, N.C.; Kouprina, N.; Kim, J.H.; Kagansky, A.; Bates, S.; Trepel, J.B.; Pommier, Y.; Sackett, D.; Larionov, V. Effects of anticancer drugs on chromosome instability and new clinical implications for tumor-suppressing therapies. Cancer Res. 2016, 76, 902–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.K.; Lovly, C.M.; Piwnica-Worms, H. Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation. Mol. Cancer Res. 2003, 1, 598–609. [Google Scholar] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Compton, D.A.; Powell, S.N.; Bastians, H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer 2017, 3, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Ward, I.M.; Wu, X.; Chen, J. Threonine 68 of Chk2 is phosphorylated at sites of DNA strand breaks. J. Biol. Chem. 2001, 276, 47755–47758. [Google Scholar] [CrossRef] [Green Version]
- Carloni, V.; Lulli, M.; Madiai, S.; Mello, T.; Hall, A.; Luong, T.V.; Pinzani, M.; Rombouts, K.; Galli, A. CHK2 overexpression and mislocalisation within mitotic structures enhances chromosomal instability and hepatocellular carcinoma progression. Gut 2018, 67, 348–361. [Google Scholar] [CrossRef]
- Tahmasebi-Birgani, M.; Ansari, H.; Carloni, V. Defective mitosis-linked DNA damage response and chromosomal instability in liver cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 60–65. [Google Scholar] [CrossRef]
- Weaver, B.A.A.; Silk, A.D.; Cleveland, D.W. Low rates of aneuploidy promote tumorigenesis while high rates of aneuploidy cause cell death and tumor suppression. Cell Oncol. 2008, 30, 453. [Google Scholar] [CrossRef]
- Janssen, A.; Kops, G.J.P.L.; Medema, R.H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad.Sci. USA 2009, 106, 19108–19113. [Google Scholar] [CrossRef] [Green Version]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.L.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Wedge, D.C.; CAMCAP Study Group; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018, 33, 125–136.e3. [Google Scholar] [CrossRef]
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [Green Version]
- Tarsounas, M.; Sung, P. The anti-tumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 2, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in BRCA1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif 1to control 5’ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–96539. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell 2018, 173, 972–988.e923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.; Peng, T.-N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by BRCA1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, A.; Hartlerode, A.; Stucki, M.; Odate, S.; Puget, N.; Kwok, A.; Nagaraju, G.; Yan, C.; Alt, F.W.; Chen, J.; et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell 2007, 28, 1045–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001, 1, 3237–3242. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.S.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary non polyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Price, D.B.; D’Andrea, D.A. Chromatin remodeling at DNA double-strand breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster-Bockler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef]
- Sasaki, S.; Mello, C.C.; Shimada, A.; Nakatani, Y.; Hashimoto, S.; Ogawa, M.; Matsushima, K.; Gu, S.G.; Kasahara, M.; Ahsan, B.; et al. Chromatin associated periodicity in genetic variation downstream of transcriptional start sites. Science 2009, 323, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolstorukov, M.Y.; Volfovsky, N.; Stephens, R.M.; Park, P.J. Impact of chromatin structure on sequence variability in the human genome. Nat. Struct. Mol. Biol. 2011, 18, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Löbrich, M. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.F.; Yeong, F.M. Safeguarding entry into mitosis: The antephase checkpoint. Mol. Cell. Biol. 2010, 30, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Deckbar, D.; Birraux, J.; Krempler, A.; Tchouandong, L.; Beucher, A.; Walker, S.; Stiff, T.; Jeggo, P.; Lobrich, M. Chromosome breakage after G2 checkpoint release. J. Cell Biol. 2007, 176, 749–755. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal 2010, 3, rs3. [Google Scholar] [CrossRef]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability an evolving hallmark of cancer. Nature Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Lulli, M.; Del Coco, L.; Mello, T.; Sukowati, C.; Madiai, S.; Gragnani, L.; Forte, P.; Fanizzi, F.P.; Mazzocca, A.; Rombouts, K.; et al. DNA damage response protein CHK2 regulates metabolism in liver cancer. Cancer Res. 2021, 81, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 2010, 10, 123–130. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16mediates NF-kB signaling after nuclear DNA damage. Mol. Cell 2018, 71, 745–760.e745. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon β-responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef] [Green Version]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.J.; Barber, G.N. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018, 37, 2037–2051. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef] [Green Version]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntington, N.D.; Cursons, J.; Rautela, J. The cancer natural killer cell immunity cycle. Nature Rev. Cancer 2020, 20, 437–454. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D. Interferon-inducible effector mechanisms in cell autonomous immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef]
- Kim, C.G.; Kim, C.; Yoon, S.E.; Kim, K.H.; Choi, S.J.; Kang, B.; Kim, H.R.; Park, S.-H.; Shin, E.-C.; Kim, Y.-Y.; et al. Hyperprogressive disease during PD-1 blockade in patients with advanced hepatocellular carcinoma. J. Hepatol. 2021, 74, 350–359. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Pharm, M.; Skora, A.D.; Luber, B.S.; Azad, N.S. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion and deletion derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M., Jr.; James, R.C.; Stetson, D.B. Human DNA-PK activates a STING-independent. DNA sensing pathway. Sci. Immunol. 2020, 5, eaba4219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reislander, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP inhibition elicits STING-dependent antitumor immunity in BRCA1-deficient ovarian cancer. Cell Rep. 2018, 25, 2972–2980.e2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, L.A. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef]
- Reislander, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol Cell. 2020, 80, 21–28. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carloni, V.; Morganti, E.; Galli, A.; Mazzocca, A. The Adaptability of Chromosomal Instability in Cancer Therapy and Resistance. Int. J. Mol. Sci. 2023, 24, 245. https://doi.org/10.3390/ijms24010245
Carloni V, Morganti E, Galli A, Mazzocca A. The Adaptability of Chromosomal Instability in Cancer Therapy and Resistance. International Journal of Molecular Sciences. 2023; 24(1):245. https://doi.org/10.3390/ijms24010245
Chicago/Turabian StyleCarloni, Vinicio, Elisa Morganti, Andrea Galli, and Antonio Mazzocca. 2023. "The Adaptability of Chromosomal Instability in Cancer Therapy and Resistance" International Journal of Molecular Sciences 24, no. 1: 245. https://doi.org/10.3390/ijms24010245
APA StyleCarloni, V., Morganti, E., Galli, A., & Mazzocca, A. (2023). The Adaptability of Chromosomal Instability in Cancer Therapy and Resistance. International Journal of Molecular Sciences, 24(1), 245. https://doi.org/10.3390/ijms24010245