
Functional Heterodimerization between the G Protein-Coupled Receptor GPR17 and the Chemokine Receptors 2 and 4: New Evidence
 , , , , , , , , and
, , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Physical Interaction between GPR17 and CXCR2 Receptor
2.2. Physical Interaction between GPR17 and CXCR4 Receptor
2.3. Functional Interaction between GPR17 and CXCR2 Receptor
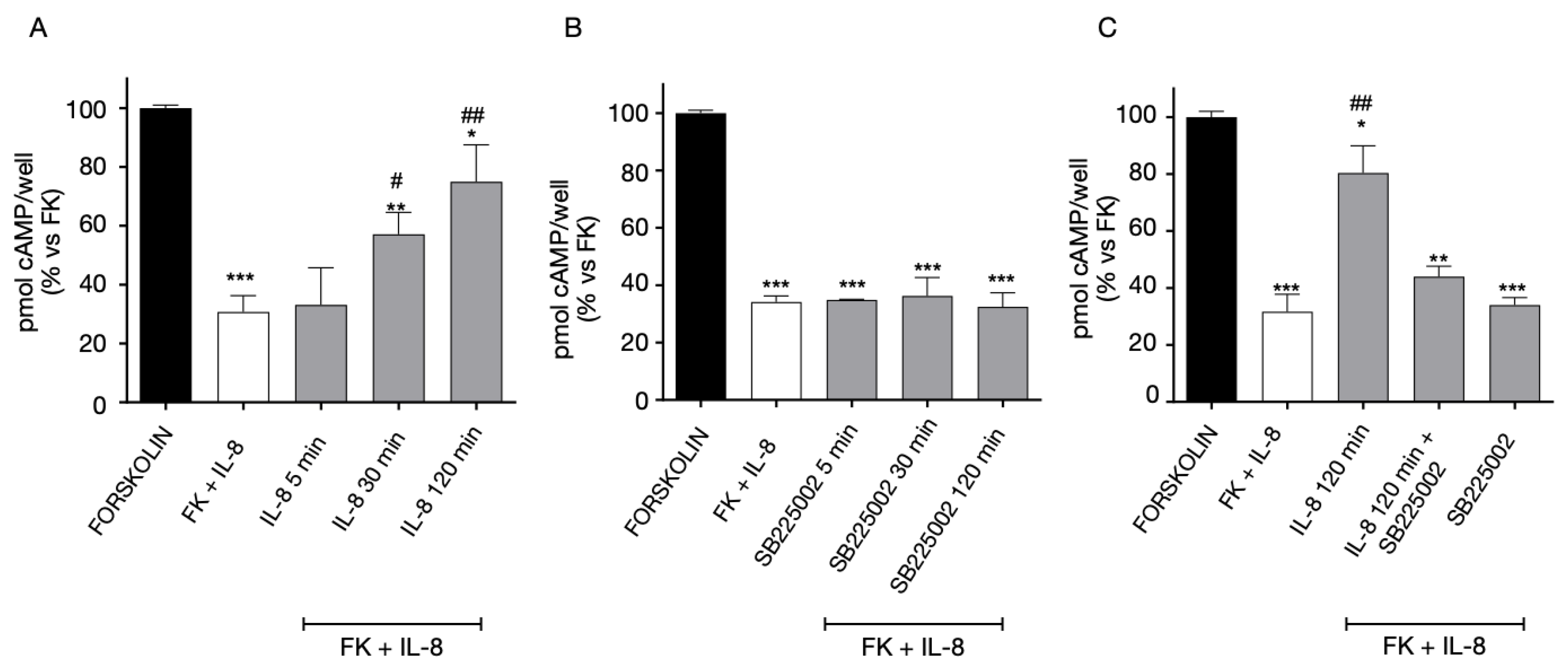
2.4. Functional Interaction between GPR17 and CXCR4 Receptor
2.5. In Silico Homology Modeling and Molecular Dynamics Simulations
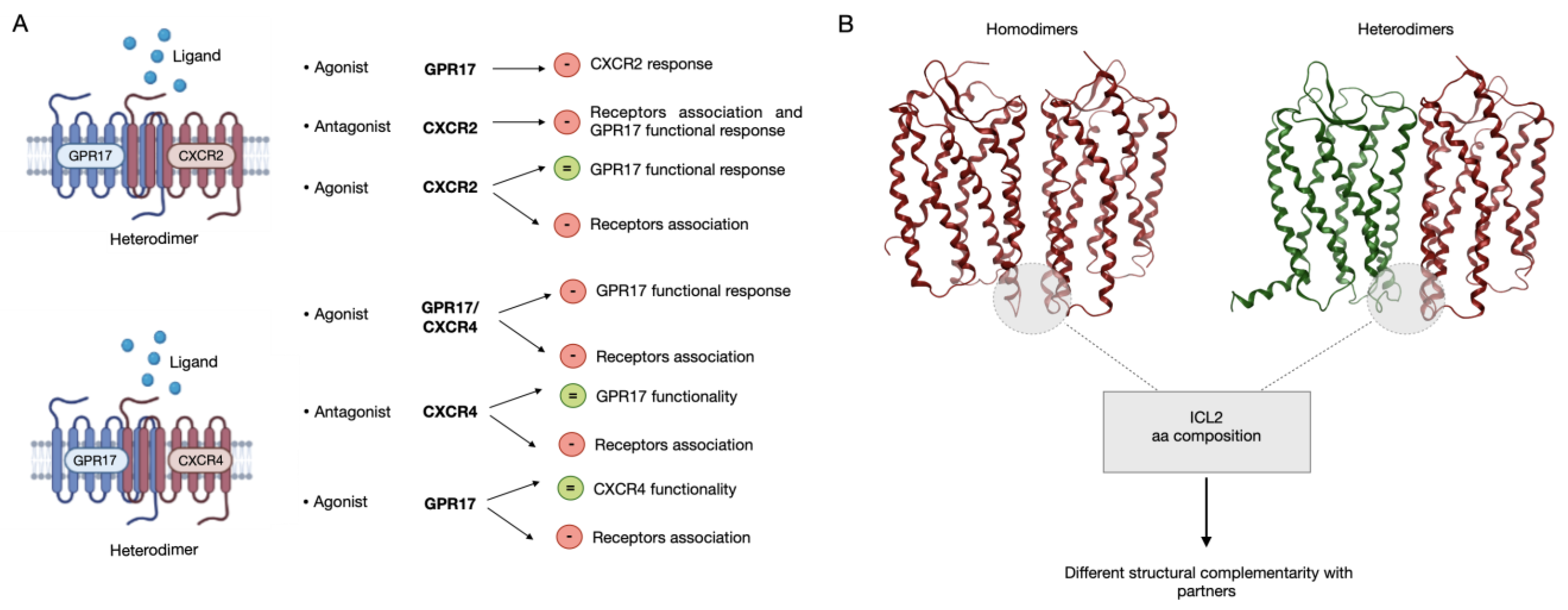
2.6. Analysis of the Dimerization Interface
2.7. Dimer Formation Energy Analysis
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Cell Lines and Culture
4.3. Plasmid Construction
4.4. Co-Immunoprecipitation-Western Blot Assay
4.5. ELISA Assay
4.6. cAMP Assay
4.7. Data Analysis
4.8. Homology Modeling Procedures
4.9. Molecular Dynamics Simulations
4.10. Dimer Formation Energy Calculation
4.11. Plotting Procedures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lecca, D.; Trincavelli, M.L.; Gelosa, P.; Sironi, L.; Ciana, P.; Fumagalli, M.; Villa, G.; Verderio, C.; Grumelli, C.; Guerrini, U.; et al. The Recently Identified P2Y-Like Receptor GPR17 Is a Sensor of Brain Damage and a New Target for Brain Repair. PLoS ONE 2008, 3, e3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziedzic, A.; Miller, E.; Saluk-Bijak, J.; Bijak, M. The Gpr17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, K.; Meren, N.; Schröder, R.; Hennen, S.; Preis, P.; Schmitt, N.K.; Peters, L.; Schrage, R.; Vermeiren, C.; Gillard, M.; et al. The Orphan Receptor GPR17 Is Unresponsive to Uracil Nucleotides and Cysteinyl Leukotrienes. Mol. Pharmacol. 2017, 91, 518–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecca, D.; Raffaele, S.; Abbracchio, M.P.; Fumagalli, M. Regulation and Signaling of the GPR17 Receptor in Oligodendroglial Cells. Glia 2020, 68, 1957–1967. [Google Scholar] [CrossRef]
- Benned-Jensen, T.; Rosenkilde, M.M. Distinct Expression and Ligand-Binding Profiles of Two Constitutively Active GPR17 Splice Variants. Br. J. Pharmacol. 2010, 159, 1092. [Google Scholar] [CrossRef] [Green Version]
- Ciana, P.; Fumagalli, M.; Trincavelli, M.L.; Verderio, C.; Rosa, P.; Lecca, D.; Ferrario, S.; Parravicini, C.; Capra, V.; Gelosa, P.; et al. The Orphan Receptor GPR17 Identified as a New Dual Uracil Nucleotides/Cysteinyl-Leukotrienes Receptor. EMBO J. 2006, 25, 4615–4627. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P. CNS Remyelination as a Novel Reparative Approach to Neurodegenerative Diseases: The Roles of Purinergic Signaling and the P2Y-like Receptor GPR17. Neuropharmacology 2016, 104, 82–93. [Google Scholar] [CrossRef]
- Fumagalli, M.; Daniele, S.; Lecca, D.; Lee, P.R.; Parravicini, C.; Fields, R.D.; Rosa, P.; Antonucci, F.; Verderio, C.; Trincavelli, M.L.; et al. Phenotypic Changes, Signaling Pathway, and Functional Correlates of GPR17-Expressing Neural Precursor Cells during Oligodendrocyte Differentiation. J. Biol. Chem. 2011, 286, 10593–10604. [Google Scholar] [CrossRef] [Green Version]
- Boda, E.; Viganò, F.; Rosa, P.; Fumagalli, M.; Labat-Gest, V.; Tempia, F.; Abbracchio, M.P.; Dimou, L.; Buffo, A. The GPR17 Receptor in NG2 Expressing Cells: Focus on in Vivocell Maturation and Participation in Acute Trauma and Chronic Damage. Glia 2011, 59, 1958–1973. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, H.; Wang, S.; Koito, H.; Li, J.; Ye, F.; Hoang, J.; Escobar, S.S.; Gow, A.; Arnett, H.A.; et al. The Oligodendrocyte-Specific G Protein–Coupled Receptor GPR17 Is a Cell-Intrinsic Timer of Myelination. Nat. Neurosci. 2009, 12, 1398–1406. [Google Scholar] [CrossRef]
- Crociara, P.; Parolisi, R.; Conte, D.; Fumagalli, M.; Bonfanti, L. Cellular and Molecular Characterization of Multipolar Map5-Expressing Cells: A Subset of Newly Generated, Stage-Specific Parenchymal Cells in the Mammalian Central Nervous System. PLoS ONE 2013, 8, e63258. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a Common Denominator for Neuroinflammation, Blood-Brain Barrier Alteration, and Oligodendrocyte Precursor Response in EAE, Plays a Role in Dendritic Cell Activation. Acta Neuropathol. 2016, 132, 23–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitew, S.; Hay, C.M.; Peckham, H.; Xiao, J.; Koenning, M.; Emery, B. Mechanisms Regulating the Development of Oligodendrocytes and Central Nervous System Myelin. Neuroscience 2014, 276, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, H.; Martin, E.; Hassani, H.; Clavairoly, A.; Maire, C.L.; Viadieu, A.; Kerninon, C.; Delmasure, A.; Frah, M.; Weber, M.; et al. Ascl1/Mash1 Promotes Brain Oligodendrogenesis during Myelination and Remyelination. J. Neurosci. 2013, 33, 9752–9768. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Bonfanti, E.; Daniele, S.; Zappelli, E.; Lecca, D.; Martini, C.; Trincavelli, M.L.; Abbracchio, M.P. The Ubiquitin Ligase Mdm2 Controls Oligodendrocyte Maturation by Intertwining MTOR with G Protein-Coupled Receptor Kinase 2 in the Regulation of GPR17 Receptor Desensitization. Glia 2015, 63, 2327–2339. [Google Scholar] [CrossRef]
- Daniele, S.; Trincavelli, M.L.; Gabelloni, P.; Lecca, D.; Rosa, P.; Abbracchio, M.P.; Martini, C. Agonist-Induced Desensitization/Resensitization of Human G Protein-Coupled Receptor 17: A Functional Cross-Talk between Purinergic and Cysteinyl-Leukotriene Ligands. J. Pharmacol. Exp. Ther. 2011, 338, 559–567. [Google Scholar] [CrossRef]
- Daniele, S.; Trincavelli, M.L.; Fumagalli, M.; Zappelli, E.; Lecca, D.; Bonfanti, E.; Campiglia, P.; Abbracchio, M.P.; Martini, C. Does GRK-β Arrestin Machinery Work as a “Switch on” for GPR17-Mediated Activation of Intracellular Signaling Pathways? Cell. Signal. 2014, 26, 1310–1325. [Google Scholar] [CrossRef]
- Sensi, C.; Daniele, S.; Parravicini, C.; Zappelli, E.; Russo, V.; Trincavelli, M.L.; Martini, C.; Abbracchio, M.P.; Eberini, I. Oxysterols Act as Promiscuous Ligands of Class-A GPCRs: In Silico Molecular Modeling and in Vitro Validation. Cell. Signal. 2014, 26, 2614–2620. [Google Scholar] [CrossRef]
- Arimitsu, N.; Shimizu, J.; Fujiwara, N.; Takai, K.; Takada, E.; Kono, T.; Ueda, Y.; Suzuki, T.; Suzuki, N. Role of SDF1/CXCR4 Interaction in Experimental Hemiplegic Models with Neural Cell Transplantation. Int. J. Mol. Sci. 2012, 13, 2636–2649. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Liu, H.; Zhang, X.; Xu, F.; Qin, L.; Cheng, P.; Huang, H. Inhibition of SDF-1α/CXCR4 Signalling in Subchondral Bone Attenuates Post-Traumatic Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 943. [Google Scholar] [CrossRef]
- Wang, R.Y.; Yang, Y.R.; Chang, H.C. The SDF1-CXCR4 Axis Is Involved in the Hyperbaric Oxygen Therapy-Mediated Neuronal Cells Migration in Transient Brain Ischemic Rats. Int. J. Mol. Sci. 2022, 23, 1780. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Fleszar, M.G.; Fortuna, P.; Gostomska-Pampuch, K.; Lewandowski, Ł.; Piasecki, T.; Kosyk, B.; Szeląg, A.; Trocha, M. Modulation of Prostanoids Profile and Counter-Regulation of Sdf-1α/Cxcr4 and Vip/Vpac2 Expression by Sitagliptin in Non-Diabetic Rat Model of Hepatic Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 13155. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sánchez, A.V.; García-España, A.; Sánchez-Gómez, P.; Font-De-mora, J.; Merino, M.; Mullor, J.L. The Embryonic Key Pluripotent Factor NANOG Mediates Glioblastoma Cell Migration via the SDF1/CXCR4 Pathway. Int. J. Mol. Sci. 2021, 22, 10620. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, C.; Daniele, S.; Palazzolo, L.; Trincavelli, M.L.; Martini, C.; Zaratin, P.; Primi, R.; Coppolino, G.; Gianazza, E.; Abbracchio, M.P.; et al. A Promiscuous Recognition Mechanism between GPR17 and SDF-1: Molecular Insights. Cell. Signal. 2016, 28, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, V.J.; Daminelli, S.; Schroeder, M.; Cavalli, A.; Rzepa, H. Drug Promiscuity in PDB: Protein Binding Site Similarity Is Key. PLoS ONE 2013, 8, e65894. [Google Scholar] [CrossRef]
- Maekawa, A.; Balestrieri, B.; Austen, K.F.; Kanaoka, Y. GPR17 Is a Negative Regulator of the Cysteinyl Leukotriene 1 Receptor Response to Leukotriene D4. Proc. Natl. Acad. Sci. USA 2009, 106, 11685–11690. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Qiao, Y.; Li, Z. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 2018, 39, 367–386. [Google Scholar] [CrossRef]
- Kasai, R.S.; Kusumi, A. Single-Molecule Imaging Revealed Dynamic GPCR Dimerization. Curr. Opin. Cell Biol. 2014, 27, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Frade, J.M.; Mellado, M.; Martínez-A, C. Chemokine Receptor Dimerization: Two Are Better than One. Trends Immunol. 2001, 22, 612–617. [Google Scholar] [CrossRef]
- Wang, J.; Norcross, M. Dimerization of Chemokine Receptors in Living Cells: Key to Receptor Function and Novel Targets for Therapy. Drug Discov. Today 2008, 13, 625–632. [Google Scholar] [CrossRef]
- Salanga, C.L.; Ohayre, M.; Handel, T. Review Modulation of Chemokine Receptor Activity through Dimerization and Crosstalk. Cell. Mol. Life Sci. 2009, 8, 1370–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.R.; O’Dowd, B.F.; Lee, S.P. G-Protein-Coupled Receptor Oligomerization and Its Potential for Drug Discovery. Nat. Rev. Drug Discov. 2002, 1, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Terrillon, S.; Bouvier, M. Roles of G-Protein-Coupled Receptor Dimerization. EMBO Rep. 2004, 5, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, G. G Protein-Coupled Receptor Hetero-Dimerization: Contribution to Pharmacology and Function. Br. J. Pharmacol. 2009, 158, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, L.M.; Holgado, B.L.; Martínez-A, C.; Rodríguez-Frade, J.M.; Mellado, M. Chemokine Receptor Oligomerization: A Further Step toward Chemokine Function. Immunol. Lett. 2012, 145, 23–29. [Google Scholar] [CrossRef]
- Eckardt, V.; Miller, M.C.; Blanchet, X.; Duan, R.; Leberzammer, J.; Duchene, J.; Soehnlein, O.; Megens, R.T.; Ludwig, A.; Dregni, A.; et al. Chemokines and Galectins Form Heterodimers to Modulate Inflammation. EMBO Rep. 2020, 21, e47852. [Google Scholar] [CrossRef]
- Song, Z.-Y.; Wang, F.; Cui, S.-X.; Gao, Z.-H.; Qu, X.-J. CXCR7/CXCR4 Heterodimer-Induced Histone Demethylation: A New Mechanism of Colorectal Tumorigenesis. Oncogene 2018, 38, 1560–1575. [Google Scholar] [CrossRef]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Raccosta, L.; Fontana, R.; Maggioni, D.; Lanterna, C.; Villablanca, E.J.; Paniccia, A.; Musumeci, A.; Chiricozzi, E.; Trincavelli, M.L.; Daniele, S.; et al. The Oxysterol-CXCR2 Axis Plays a Key Role in the Recruitment of Tumor-Promoting Neutrophils. J. Exp. Med. 2013, 210, 1711–1728. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.M.; Pridgen, B.C.; Haribabu, B.; Ali, H.; Snyderman, R. Differential Cross-Regulation of the Human Chemokine Receptors CXCR1 and CXCR2. Evidence for Time-Dependent Signal Generation. J. Biol. Chem. 1998, 273, 23830–23836. [Google Scholar] [CrossRef]
- Nasser, M.W.; Raghuwanshi, S.K.; Malloy, K.M.; Gangavarapu, P.; Shim, J.Y.; Rajarathnam, K.; Richardson, R.M. CXCR1 and CXCR2 Activation and Regulation: Role of Aspartate 199 of the Second Extracellular Loop of CXCR2 in CXCL8-Mediated Rapid Receptor Internalization. J. Biol. Chem. 2007, 282, 6906–6915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, T.J. Bidirectional Regulation of Opioid and Chemokine Function. Front. Immunol. 2020, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Wong, T.-S.; Chen, G.; Zhang, Z.; Zhang, B.; Gan, S.; Gao, W.; Li, J.; Wu, Z.; Pan, X.; et al. Cryo-EM Structure of G-Protein-Coupled Receptor GPR17 Incomplex with Inhibitory G Protein. Med. Comm. 2022, 4, e159. [Google Scholar]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Madan Babu, M. Molecular Signatures of G-Protein-Coupled Receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, J.; Navarro, G.; Casadó-Anguera, V.; Azdad, K.; Rea, W.; Moreno, E.; Brugarolas, M.; Mallol, J.; Canela, E.I.; Lluís, C.; et al. Allosteric Interactions between Agonists and Antagonists within the Adenosine A2A Receptor-Dopamine D2 Receptor Heterotetramer. Proc. Natl. Acad. Sci. USA 2015, 112, E3609–E3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trincavelli, M.L.; Cuboni, S.; Dell’Osso, M.C.; Maggio, R.; Klotz, K.N.; Novi, F.; Panighini, A.; Daniele, S.; Martini, C. Receptor Crosstalk: Haloperidol Treatment Enhances A2A Adenosine Receptor Functioning in a Transfected Cell Model. Purinergic Signal. 2010, 6, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Jordan, B.A.; Devi, L.A. G-Protein-Coupled Receptor Heterodimerization Modulates Receptor Function. Nature 1999, 399, 697–700. [Google Scholar] [CrossRef]
- Said, A.; Lother, H.; Quitterer, U. AT1-Receptor Heterodimers Show Enhanced G-Protein Activation and Altered Receptor Sequestration. Nature 2000, 407, 94–98. [Google Scholar]
- George, S.R.; Fan, T.; Xie, Z.; Tse, R.; Tam, V.; Varghese, G.; O’Dowd, B.F. Oligomerization of Mu- and Delta-Opioid Receptors. Generation of Novel Functional Properties. J. Biol. Chem. 2000, 275, 26128–26135. [Google Scholar] [CrossRef] [Green Version]
- Gomes, I.; Jordan, B.A.; Gupta, A.; Trapaidze, N.; Nagy, V.; Devi, L.A. Heterodimerization of μ and δ Opioid Receptors: A Role in Opiate Synergy. J. Neurosci. 2000, 20, RC110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocheville, M.; Lange, D.C.; Kumar, U.; Patel, S.C.; Patel, R.C.; Patel, Y.C. Receptors for Dopamine and Somatostatin: Formation of Hetero-Oligomers with Enhanced Functional Activity. Science 2000, 288, 154–157. [Google Scholar] [CrossRef] [PubMed]
- González-Maeso, J.; Ang, R.L.; Yuen, T.; Chan, P.; Weisstaub, N.V.; López-Giménez, J.F.; Zhou, M.; Okawa, Y.; Callado, L.F.; Milligan, G.; et al. Identification of a Serotonin/Glutamate Receptor Complex Implicated in Psychosis. Nature 2008, 452, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, R.; Devi, L.A. Exploring a Role for Heteromerization in GPCR Signalling Specificity. Biochem. J. 2011, 433, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.; Bailey, C.P.; Henderson, G. Agonist-Selective Mechanisms of GPCR Desensitization. Br. J. Pharmacol. 2008, 153 (Suppl. S1), S379–S388. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Shi, J.; Zemaitaitis, B.W.; Muma, N.A. Olanzapine Increases RGS7 Protein Expression via Stimulation of the Janus Tyrosine Kinase-Signal Transducer and Activator of Transcription Signaling Cascade. J. Pharmacol. Exp. Ther. 2007, 322, 133–140. [Google Scholar] [CrossRef]
- Neve, K.A.; Molinoff, P.B. Effects of Chronic Administration of Agonists and Antagonists on the Density of Beta-Adrenergic Receptors. Am. J. Cardiol. 1986, 57, F17–F22. [Google Scholar] [CrossRef]
- Nasser, M.W.; Marjoram, R.J.; Brown, S.L.; Richardson, R.M. Cross-Desensitization among CXCR1, CXCR2, and CCR5: Role of Protein Kinase C-Epsilon. J. Immunol. 2005, 174, 6927–6933. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Albee, L.J.; Volkman, B.F.; Gaponenko, V.; Majetschak, M. Asymmetrical Ligand-Induced Cross-Regulation of Chemokine (C-X-C Motif) Receptor 4 by A1-Adrenergic Receptors at the Heteromeric Receptor Complex. Sci. Reports 2018, 8, 2730. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Enten, G.A.; DeSantis, A.J.; Volkman, B.F.; Gaponenko, V.; Majetschak, M. Characterization of Heteromeric Complexes between Chemokine (C-X-C Motif) Receptor 4 and α 1-Adrenergic Receptors Utilizing Intermolecular Bioluminescence Resonance Energy Transfer Assays. Biochem. Biophys. Res. Commun. 2020, 528, 368–375. [Google Scholar] [CrossRef]
- Rodríguez, D.; Gutiérrez-de-Terán, H. Characterization of the Homodimerization Interface and Functional Hotspots of the CXCR4 Chemokine Receptor. Proteins Struct. Funct. Bioinforma. 2012, 80, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Marsango, S.; Jolly, R.; Stoneman, M.R.; Biener, G.; Handel, T.M.; Raicu, V.; Milligan, G. Chemokine Receptor CXCR4 Oligomerization Is Disrupted Selectively by the Antagonist Ligand IT1t. J. Biol. Chem. 2021, 296, 100139. [Google Scholar] [CrossRef] [PubMed]
- Angelini, J.; Marangon, D.; Raffaele, S.; Lecca, D.; Abbracchio, M.P. The Distribution of GPR17-Expressing Cells Correlates with White Matter Inflammation Status in Brain Tissues of Multiple Sclerosis Patients. Int. J. Mol. Sci. 2021, 22, 4574. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, E.; Bonifacino, T.; Raffaele, S.; Milanese, M.; Morgante, E.; Bonanno, G.; Abbracchio, M.P.; Fumagalli, M. Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice. Int. J. Mol. Sci. 2020, 21, 2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfanti, E.; Gelosa, P.; Fumagalli, M.; Dimou, L.; Viganò, F.; Tremoli, E.; Cimino, M.; Sironi, L.; Abbracchio, M.P. The Role of Oligodendrocyte Precursor Cells Expressing the GPR17 Receptor in Brain Remodeling after Stroke. Cell Death Dis. 2017, 8, e2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capelli, D.; Parravicini, C.; Pochetti, G.; Montanari, R.; Temporini, C.; Rabuffetti, M.; Trincavelli, M.L.; Daniele, S.; Fumagalli, M.; Saporiti, S.; et al. Surface Plasmon Resonance as a Tool for Ligand Binding Investigation of Engineered GPR17 Receptor, a G Protein Coupled Receptor Involved in Myelination. Front. Chem. 2020, 7, 910. [Google Scholar] [CrossRef]
- White, J.R.; Lee, J.M.; Young, P.R.; Hertzberg, R.P.; Jurewicz, A.J.; Chaikin, M.A.; Widdowson, K.; Foley, J.J.; Martin, L.D.; Griswold, D.E.; et al. Identification of a Potent, Selective Non-Peptide CXCR2 Antagonist That Inhibits Interleukin-8-Induced Neutrophil Migration. J. Biol. Chem. 1998, 273, 10095–10098. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Rettig, M.P.; Cashen, A.F. Plerixafor, a CXCR4 Antagonist for the Mobilization of Hematopoietic Stem Cells. Expert Opin. Biol. Ther. 2008, 8, 1797–1804. [Google Scholar] [CrossRef]
- Zappelli, E.; Daniele, S.; Vergassola, M.; Ceccarelli, L.; Chelucci, E.; Mangano, G.; Durando, L.; Ragni, L.; Martini, C. A Specific Combination of Nutraceutical Ingredients Exerts Cytoprotective Effects in Human Cholinergic Neurons. PharmaNutrition 2022, 22, 100317. [Google Scholar] [CrossRef]
- Zappelli, E.; Daniele, S.; Abbracchio, M.P.; Martini, C.; Trincavelli, M.L. A Rapid and Efficient Immunoenzymatic Assay to Detect Receptor Protein Interactions: G Protein-Coupled Receptors. Int. J. Mol. Sci. 2014, 15, 6252–6264. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Taliani, S.; Da Pozzo, E.; Giacomelli, C.; Costa, B.; Trincavelli, M.L.; Rossi, L.; La Pietra, V.; Barresi, E.; Carotenuto, A.; et al. Apoptosis Therapy in Cancer: The First Single-Molecule Co-Activating P53 and the Translocator Protein in Glioblastoma. Sci. Rep. 2014, 4, 4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.M.; Di Tommaso, P.; Taly, J.F.; Notredame, C. Accurate Multiple Sequence Alignment of Transmembrane Proteins with PSI-Coffee. BMC Bioinform. 2012, 13, S1. [Google Scholar] [CrossRef] [PubMed]
- Braune, M.; Scherf, N.; Heine, C.; Sygnecka, K.; Pillaiyar, T.; Parravicini, C.; Heimrich, B.; Abbracchio, M.P.; Müller, C.E.; Franke, H. Involvement of GPR17 in Neuronal Fibre Outgrowth. Int. J. Mol. Sci. 2021, 22, 11683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, Z.G.; Zhang, K.; Kiselev, E.; Crane, S.; Wang, J.; Paoletta, S.; Yi, C.; Ma, L.; Zhang, W.; et al. Two Disparate Ligand Binding Sites in the Human P2Y1 Receptor. Nature 2015, 520, 317. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.J. Structural Basis of CXC Chemokine Receptor 2 Activation and Signalling. Nature 2020, 585, 135–140. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; Van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chemie-Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 92–96. [Google Scholar]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daniele, S.; Saporiti, S.; Capaldi, S.; Pietrobono, D.; Russo, L.; Guerrini, U.; Laurenzi, T.; Ataie Kachoie, E.; Palazzolo, L.; Russo, V.; et al. Functional Heterodimerization between the G Protein-Coupled Receptor GPR17 and the Chemokine Receptors 2 and 4: New Evidence. Int. J. Mol. Sci. 2023, 24, 261. https://doi.org/10.3390/ijms24010261
Daniele S, Saporiti S, Capaldi S, Pietrobono D, Russo L, Guerrini U, Laurenzi T, Ataie Kachoie E, Palazzolo L, Russo V, et al. Functional Heterodimerization between the G Protein-Coupled Receptor GPR17 and the Chemokine Receptors 2 and 4: New Evidence. International Journal of Molecular Sciences. 2023; 24(1):261. https://doi.org/10.3390/ijms24010261
Chicago/Turabian StyleDaniele, Simona, Simona Saporiti, Stefano Capaldi, Deborah Pietrobono, Lara Russo, Uliano Guerrini, Tommaso Laurenzi, Elham Ataie Kachoie, Luca Palazzolo, Vincenzo Russo, and et al. 2023. "Functional Heterodimerization between the G Protein-Coupled Receptor GPR17 and the Chemokine Receptors 2 and 4: New Evidence" International Journal of Molecular Sciences 24, no. 1: 261. https://doi.org/10.3390/ijms24010261