
Salicylate Sodium Suppresses Monocyte Chemoattractant Protein-1 Production by Directly Inhibiting Phosphodiesterase 3B in TNF-α-Stimulated Adipocytes


Abstract
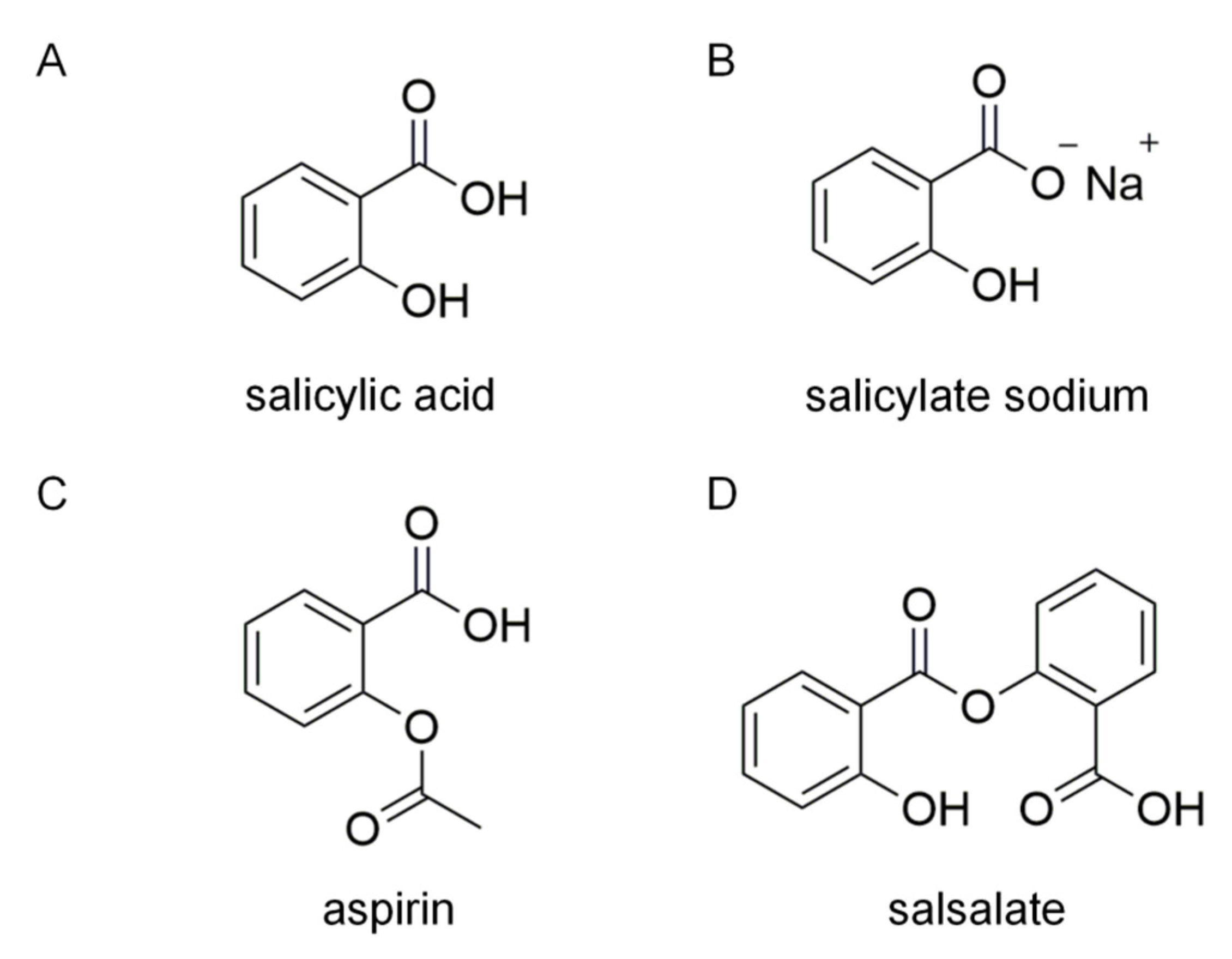
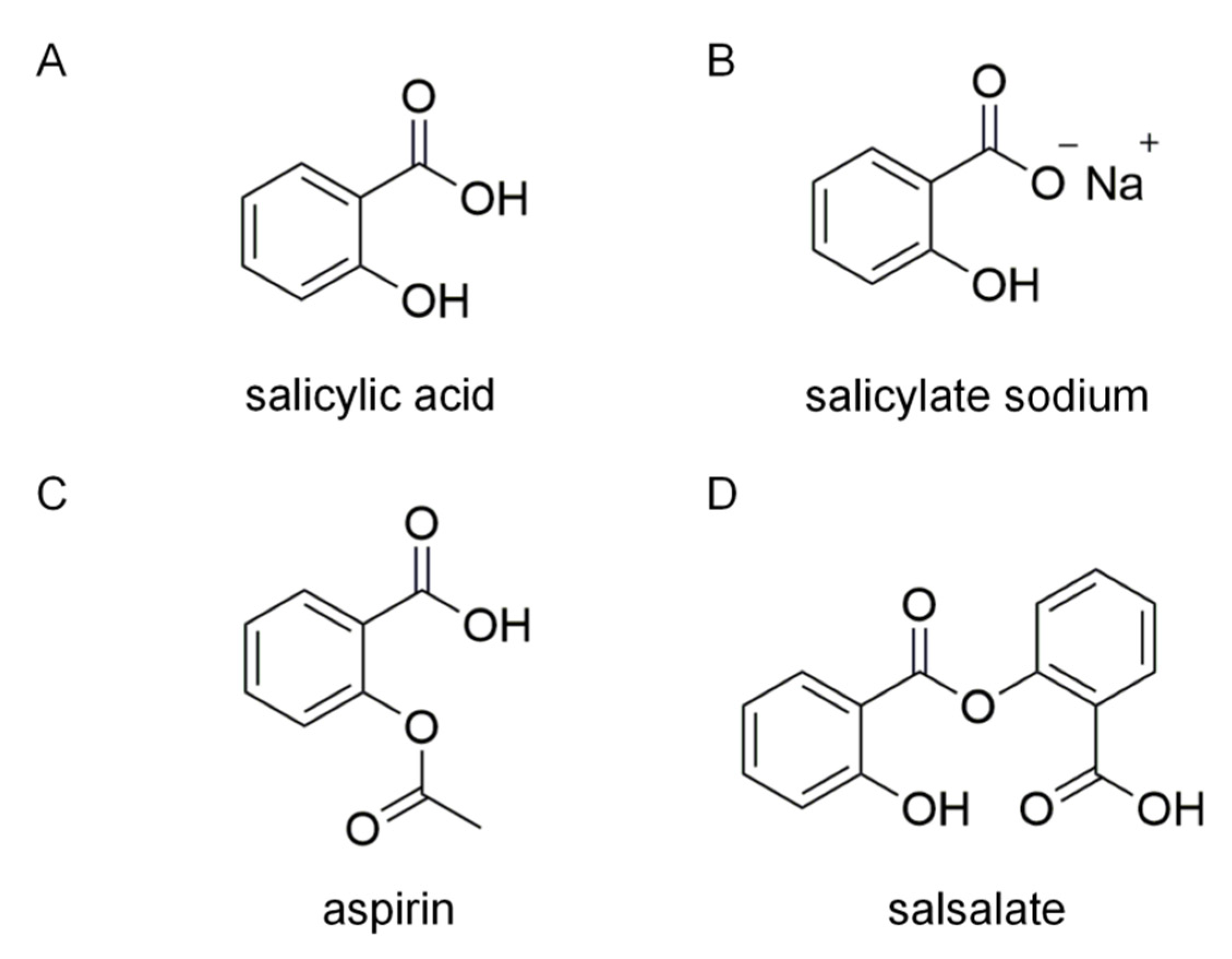
:1. Introduction
2. Results
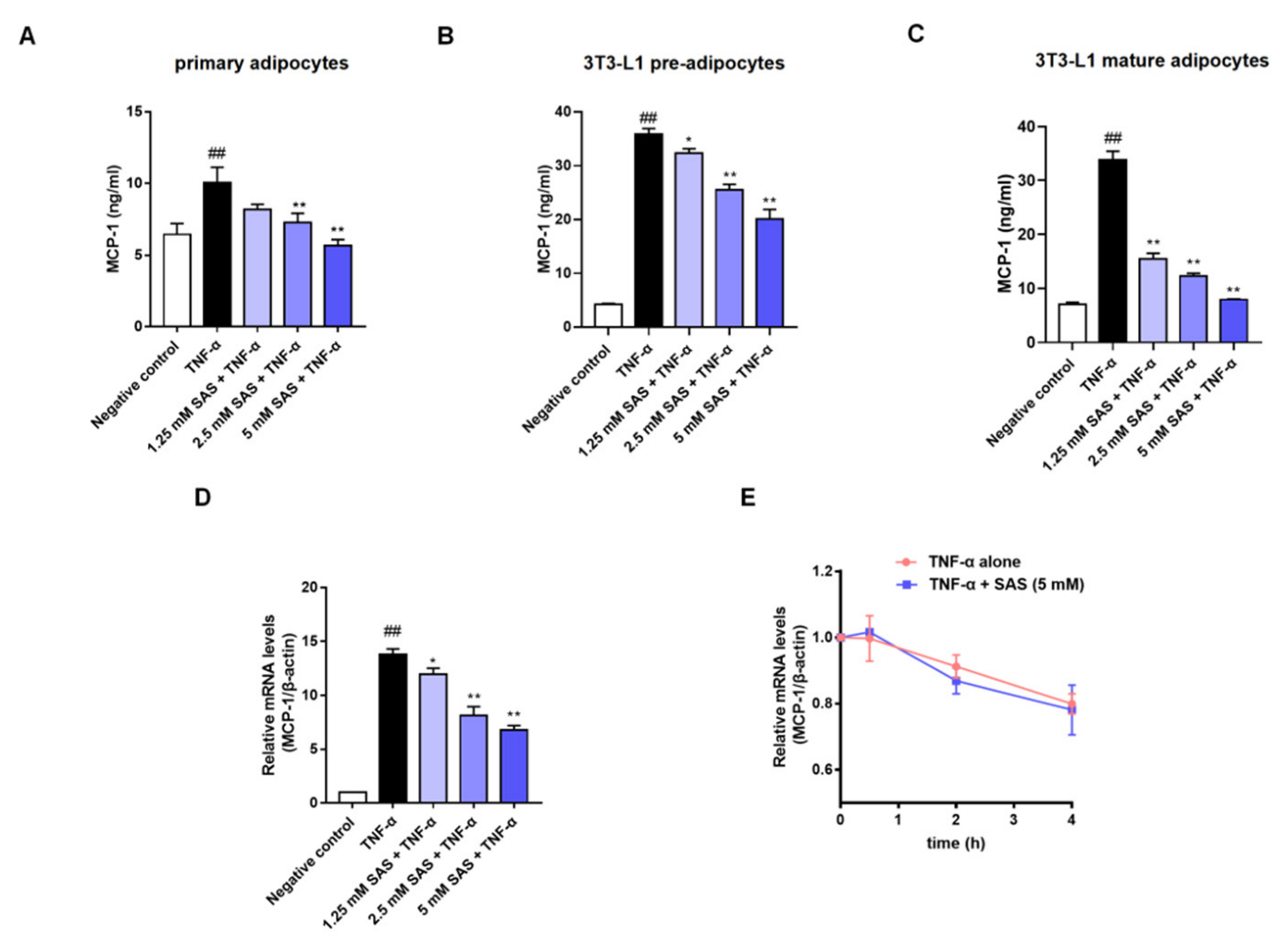
2.1. SAS Decreases MCP-1 Production in TNF-α-Stimulated Adipocytes In Vitro
2.2. SAS Decreases MCP-1 Production Independent of IKKβ Inhibition and AMPK Activation in Adipocytes
2.3. SAS Reduces the Levels of p-p38 and p-ERK and Increases MKP-1 Expression in TNF-α-Stimulated Adipocytes
2.4. SAS Decreases MCP-1 Production Depending on cAMP/PKA Pathway in Adipocytes
2.5. SAS Directly Inhibits PDE3B to Decrease MCP-1 Production in Adipocytes
2.6. SAS Is a Non-Specific PDE3B Inhibitor
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cells and Animals
4.3. Isolation and Culture of Mouse Primary Mature Adipocytes
4.4. Differentiation of 3T3-L1 Adipocytes
4.5. Determination of MCP-1 Level in Supernatant or Subnatant
4.6. RNA Extraction and Quantitative Real-Time PCR (RT-qPCR)
4.7. Western Blot Assay
4.8. Determination of Intracellular cAMP Level
4.9. PKA Activity Assay
4.10. Surface Plasmon Resonance (SPR) Assay
4.11. Gene Silencing
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Hammarstedt, A.; Andersson, C.X.; Smith, U. Inflamed adipose tissue: A culprit underlying the metabolic syndrome and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Chen, Q.; Shah, S.; Du, J.; Tao, B.; Tzameli, I.; Yan, W.; Xu, H. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: Involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 2009, 58, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.-I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Albert, V.; Wölnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Zhao, A.Z.; Zhao, H.; Teague, J.; Fujimoto, W.; Beavo, J.A. Attenuation of insulin secretion by insulin-like growth factor 1 is mediated through activation of phosphodiesterase 3B. Proc. Natl. Acad. Sci. USA 1997, 94, 3223–3228. [Google Scholar] [CrossRef] [Green Version]
- Shakur, Y.; Holst, L.S.; Landstrom, T.R.; Movsesian, M.; Degerman, E.; Manganiello, V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 66, 241–277. [Google Scholar] [CrossRef]
- Eriksson, J.W.; Wesslau, C.; Smith, U. The cGMP-inhibitable phosphodiesterase modulates glucose transport activation by insulin. Biochim. Biophys. Acta 1994, 1189, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Zmuda-Trzebiatowska, E.; Oknianska, A.; Manganiello, V.; Degerman, E. Role of PDE3B in insulin-induced glucose uptake, GLUT-4 translocation and lipogenesis in primary rat adipocytes. Cell Signal. 2006, 18, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.A.; Chung, Y.W.; Tang, Y.; Hockman, S.C.; Liu, S.; Khan, Y.; Huo, K.; Billings, E.; Amar, M.J.; Remaley, A.T.; et al. Phosphodiesterase 3B (PDE3B) regulates NLRP3 inflammasome in adipose tissue. Sci. Rep. 2016, 6, 28056. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Sun, Y.; Jin, X.; Wen, W.; Cao, Y. Diosgenin Inhibits Excessive Proliferation and Inflammatory Response of Synovial Fibroblasts in Rheumatoid Arthritis by Targeting PDE3B. Inflammation 2021, 44, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Beute, J.; Lukkes, M.; Koekoek, E.P.; Nastiti, H.; Ganesh, K.; De Bruijn, M.J.; Hockman, S.; Van Nimwegen, M.; Braunstahl, G.; Boon, L.; et al. A pathophysiological role of PDE3 in allergic airway inflammation. JCI Insight 2018, 3, e94888. [Google Scholar] [CrossRef] [Green Version]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Kim, Y.-J.; Fillmore, J.J.; Chen, Y.; Moore, I.; Lee, J.; Yuan, M.; Li, Z.W.; Karin, M.; Perret, P.; et al. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Investig. 2001, 108, 437–446. [Google Scholar] [CrossRef]
- Hundal, R.S.; Petersen, K.F.; Mayerson, A.B.; Randhawa, P.S.; Inzucchi, S.; Shoelson, S.E.; Shulman, G.I. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J. Clin. Investig. 2002, 109, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Möhlig, M.; Freudenberg, M.; Bobbert, T.; Ristow, M.; Rochlitz, H.; Weickert, M.O.; Pfeiffer, A.F.H.; Spranger, J. Acetylsalicylic acid improves lipid-induced insulin resistance in healthy men. J. Clin. Endocrinol. Metab. 2006, 91, 964–967. [Google Scholar] [CrossRef] [Green Version]
- Goldfine, A.B.; Fonseca, V.; Jablonski, K.A.; Pyle, L.; Staten, M.A.; Shoelson, S.E. The effects of salsalate on glycemic control in patients with type 2 diabetes: A randomized trial. Ann. Intern. Med. 2010, 152, 346–357. [Google Scholar] [CrossRef]
- Shirakawa, K.; Wang, L.; Man, N.; Maksimoska, J.; Sorum, A.W.; Lim, H.W.; Lee, I.S.; Shimazu, T.; Newman, J.C.; Schröder, S.; et al. Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. Elife. 2016, 5, e11156. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Neels, J.G.; Fan, W.; Li, P.P.; Nguyen, M.T.; Olefsky, J.M. Glucocorticoids and thiazolidinediones interfere with adipocyte-mediated macrophage chemotaxis and recruitment. J. Biol. Chem. 2009, 284, 31223–31235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, D.; Boekhoudt, G.H.; Rogers, E.M.; Boss, J.M. Nuclear factor-kappa B p65 mediates the assembly and activation of the TNF-responsive element of the murine monocyte chemoattractant-1 gene. J. Immunol. 1999, 162, 727–734. [Google Scholar] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Wang, W.; Lin, Q.; Lin, R.; Zhang, J.; Ren, F.; Zhang, J.; Ji, M.; Li, Y. PPARα agonist fenofibrate attenuates TNF-α-induced CD40 expression in 3T3-L1 adipocytes via the SIRT1-dependent signaling pathway. Exp. Cell Res. 2013, 319, 1523–1533. [Google Scholar] [CrossRef]
- Nagahara, K.; Dobashi, K.; Ishikawa, T.; Nakano, Y.; Abe, Y.; Tanaka, D.; Itabashi, K. AICAR Attenuates TNFα-Induced Inappropriate Secretion of Monocyte Chemoattractant Protein-1 and Adiponectin in 3T3-L1 Adipocytes. J. Atheroscler. Thromb. 2016, 23, 1345–1354. [Google Scholar] [CrossRef]
- Ito, A.; Suganami, T.; Miyamoto, Y.; Yoshimasa, Y.; Takeya, M.; Kamei, Y.; Ogawa, Y. Role of MAPK phosphatase-1 in the induction of monocyte chemoattractant protein-1 during the course of adipocyte hypertrophy. J. Biol. Chem. 2007, 282, 25445–25452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Zhu, N.; Yu, M.; Shen, B.; Xiang, J.; Lin, A. Cyclic AMP inhibits JNK activation by CREB-mediated induction of c-FLIP(L) and MKP-1, thereby antagonizing UV-induced apoptosis. Cell Death Differ. 2008, 15, 1654–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahn Landström, T.; Mei, J.; Karlsson, M.; Manganiello, V.; Degerman, E. Down-regulation of cyclic-nucleotide phosphodiesterase 3B in 3T3-L1 adipocytes induced by tumour necrosis factor alpha and cAMP. Biochem. J. 2000, 346, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Cook, N.R.; Gaziano, J.M.; Price, J.F.; Belch, J.F.F.; Roncaglioni, M.C.; Morimoto, T.; Mehta, Z. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: Analysis of individual patient data from randomised trials. Lancet 2018, 392, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef]
- Arkan, M.C.; Hevener, A.L.; Greten, F.; Maeda, S.; Li, Z.-W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B.Z. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef] [Green Version]
- Mottillo, E.P.; Desjardins, E.M.; Crane, J.D.; Smith, B.K.; Green, A.E.; Ducommun, S.; Henriksen, T.I.; Rebalka, I.A.; Razi, A.; Sakamoto, K.; et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016, 24, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Cho, N.H.; Seong, S.Y.; Choi, M.S.; Kim, I.S. Expression of chemokine genes in human dermal microvascular endothelial cell lines infected with Orientia tsutsugamushi. Infect. Immun. 2001, 69, 1265–1272. [Google Scholar] [CrossRef]
- Cho, N.H.; Seong, S.Y.; Huh, M.S.; Kim, N.H.; Choi, M.S.; Kim, I.S. Induction of the gene encoding macrophage chemoattractant protein 1 by Orientia tsutsugamushi in human endothelial cells involves activation of transcription factor activator protein 1. Infect. Immun. 2002, 70, 4841–4850. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jiang, X.; Deng, B.; Xiao, J.; Jin, J.; Huang, Z. Lipopolysaccharide and palmitic acid synergistically induced MCP-1 production via MAPK-meditated TLR4 signaling pathway in RAW264.7 cells. Lipids Health Dis. 2019, 18, 71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, Z.; Li, W.; Kang, Y.; Xu, Z.; Li, X.; Gao, Y.; Qi, Y. MAPKs/AP-1, not NF-κB, is responsible for MCP-1 production in TNF-α-activated adipocytes. Adipocyte 2022, 11, 477–486. [Google Scholar] [CrossRef]
- Panganiban, R.P.; Vonakis, B.M.; Ishmael, F.T.; Stellato, C. Coordinated post-transcriptional regulation of the chemokine system: Messages from CCL2. J. Interferon Cytokine Res. 2014, 34, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Charles, C.H.; Lau, L.F.; Tonks, N.K. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993, 75, 487–493. [Google Scholar] [CrossRef]
- Franklin, C.C.; Kraft, A.S. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J. Biol. Chem. 1997, 272, 16917–16923. [Google Scholar] [CrossRef] [Green Version]
- Burgun, C.; Esteve, L.; Humblot, N.; Aunis, D.; Zwiller, J. Cyclic AMP-elevating agents induce the expression of MAP kinase phosphatase-1 in PC12 cells. FEBS. Lett. 2000, 484, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Brion, L.; Maloberti, P.M.; Gomez, N.V.; Poderoso, C.; Gorostizaga, A.B.; Garcia, M.M.M.S.; Acquier, A.B.; Cooke, M.; Mendez, C.F.; Podesta, E.J.; et al. MAPK phosphatase-1 (MKP-1) expression is up-regulated by hCG/cAMP and modulates steroidogenesis in MA-10 Leydig cells. Endocrinology 2011, 152, 2665–2677. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, R.; Hömmö, T.; Keränen, T.; Laavola, M.; Hämäläinen, M.; Vuolteenaho, K.; Lehtimäki, L.; Kankaanranta, H.; Moilanen, E. Attenuation of TNF production and experimentally induced inflammation by PDE4 inhibitor rolipram is mediated by MAPK phosphatase-1. Br. J. Pharmacol. 2013, 169, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.S.; Prabhala, P.; Oliver, B.G.; Ammit, A.J. Inhibitors of Phosphodiesterase 4, but Not Phosphodiesterase 3, Increase β2-Agonist-Induced Expression of Antiinflammatory Mitogen-Activated Protein Kinase Phosphatase 1 in Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 634–640. [Google Scholar] [CrossRef]
- Keränen, T.; Hömmö, T.; Hämäläinen, M.; Moilanen, E.; Korhonen, R. Anti-Inflammatory Effects of β2-Receptor Agonists Salbutamol and Terbutaline Are Mediated by MKP-1. PLoS ONE 2016, 11, e0148144. [Google Scholar] [CrossRef] [Green Version]
- Aronoff, D.M.; Carstens, J.K.; Chen, G.H.; Toews, G.B.; Peters-Golden, M. Short communication: Differences between macrophages and dendritic cells in the cyclic AMP-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J. Interferon Cytokine Res. 2006, 26, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Wall, E.A.; Zavzavadjian, J.R.; Chang, M.S.; Randhawa, B.; Zhu, X.; Hsueh, R.C.; Liu, J.; Driver, A.; Bao, X.R.; Sternweis, P.C.; et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2009, 2, ra28. [Google Scholar] [CrossRef] [Green Version]
- Degerman, E.; Ahmad, F.; Chung, Y.W.; Guirguis, E.; Omar, B.; Stenson, L.; Manganiello, V. From PDE3B to the regulation of energy homeostasis. Curr Opin Pharmacol. 2011, 11, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Guirguis, E.; Hockman, S.; Chung, Y.W.; Ahmad, F.; Gavrilova, O.; Raghavachari, N.; Yang, Y.; Niu, G.; Chen, X.; Yu, Z.X.; et al. A role for phosphodiesterase 3B in acquisition of brown fat characteristics by white adipose tissue in male mice. Endocrinology 2013, 154, 3152–3167. [Google Scholar] [CrossRef]
- Matsunaga, H.; Hokari, R.; Higashiyama, M.; Kurihara, C.; Okada, Y.; Watanabe, C.; Komoto, S.; Nakamura, M.; Kawaguchi, A.; Nagao, S.; et al. Cilostazol, a specific PDE-3 inhibitor, ameliorates chronic ileitis via suppression of interaction of platelets with monocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1077–G1084. [Google Scholar] [CrossRef]
- Ruan, H.; Zarnowski, M.J.; Cushman, S.W.; Lodish, H.F. Standard isolation of primary adipose cells from mouse epididymal fat pads induces inflammatory mediators and down-regulates adipocyte genes. J. Biol. Chem. 2003, 278, 47585–47593. [Google Scholar] [CrossRef] [Green Version]
- Fernyhough, M.E.; Vierck, J.L.; Hausman, G.J.; Mir, P.S.; Okine, E.K.; Dodson, M.V. Primary adipocyte culture: Adipocyte purification methods may lead to a new understanding of adipose tissue growth and development. Cytotechnology 2004, 46, 163–172. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, K.; Voigt, V.; Wabitsch, M.; Brandsch, M. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem. 2012, 425, 88–90. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Zhang, M.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. Nuclear to cytoplasmic translocation of heterogeneous nuclear ribonucleoprotein U enhances TLR-induced proinflammatory cytokine production by stabilizing mRNAs in macrophages. J. Immunol. 2012, 188, 3179–3187. [Google Scholar] [CrossRef]
- Löfås, S. Biacore systems: Leading the revolution in label-free interaction analysis. Biointerphases 2008, 3, Fd2–Fd3. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Li, Y.; Zhu, Y.; Zhang, J.; Li, Y.; Liu, X.; Jiang, W.; Yu, S.; You, X.-F.; Xiao, C.; et al. Identification of antituberculosis agents that target ribosomal protein interactions using a yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 2012, 109, 17412–17417. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA Name | Strand | Sequence (5′ to 3′) |
|---|---|---|
| PDE3B-1 | sense | CCU CAU CGU UGC UGA CUA ATT |
| antisense | UUA GUC AGC AAC GAU GAG GTT | |
| PDE3B-2 | sense | CCA UUG AAC AUU GUG GAA ATT |
| antisense | UUU CCA CAA UGU UCA AUG GTT | |
| PDE3B-3 | sense | CCA UGA AAC GGA AAC CAA ATT |
| antisense | UUU GGU UUC CGU UUC AUG GTT | |
| IKKβ-1 | sense | GUG AAC AGA UCG CCA UCA ATT |
| antisense | UUG AUG GCG AUC UGU UCA CTT | |
| IKKβ-2 | sense | GGA CAU CGU UGU UAG UGA ATT |
| antisense | UUC ACU AAC AAC GAU GUC CTT | |
| Scrambled siRNA | sense | UUC UCC GAA CGU GUC ACG UTT |
| antisense | ACG UGA CAC GUU CGG AGA ATT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Gao, Y.; Liu, Z.; Li, W.; Kang, Y.; Li, X.; Xu, Z.; Peng, C.; Qi, Y. Salicylate Sodium Suppresses Monocyte Chemoattractant Protein-1 Production by Directly Inhibiting Phosphodiesterase 3B in TNF-α-Stimulated Adipocytes. Int. J. Mol. Sci. 2023, 24, 320. https://doi.org/10.3390/ijms24010320
Zhang X, Gao Y, Liu Z, Li W, Kang Y, Li X, Xu Z, Peng C, Qi Y. Salicylate Sodium Suppresses Monocyte Chemoattractant Protein-1 Production by Directly Inhibiting Phosphodiesterase 3B in TNF-α-Stimulated Adipocytes. International Journal of Molecular Sciences. 2023; 24(1):320. https://doi.org/10.3390/ijms24010320
Chicago/Turabian StyleZhang, Xiaoyu, Yuan Gao, Zhuangzhuang Liu, Wenjing Li, Yuan Kang, Ximeng Li, Zhenlu Xu, Cheng Peng, and Yun Qi. 2023. "Salicylate Sodium Suppresses Monocyte Chemoattractant Protein-1 Production by Directly Inhibiting Phosphodiesterase 3B in TNF-α-Stimulated Adipocytes" International Journal of Molecular Sciences 24, no. 1: 320. https://doi.org/10.3390/ijms24010320
APA StyleZhang, X., Gao, Y., Liu, Z., Li, W., Kang, Y., Li, X., Xu, Z., Peng, C., & Qi, Y. (2023). Salicylate Sodium Suppresses Monocyte Chemoattractant Protein-1 Production by Directly Inhibiting Phosphodiesterase 3B in TNF-α-Stimulated Adipocytes. International Journal of Molecular Sciences, 24(1), 320. https://doi.org/10.3390/ijms24010320