
Molecular Determinants of Fibrillation in a Viral Amyloidogenic Domain from Combined Biochemical and Biophysical Studies
 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
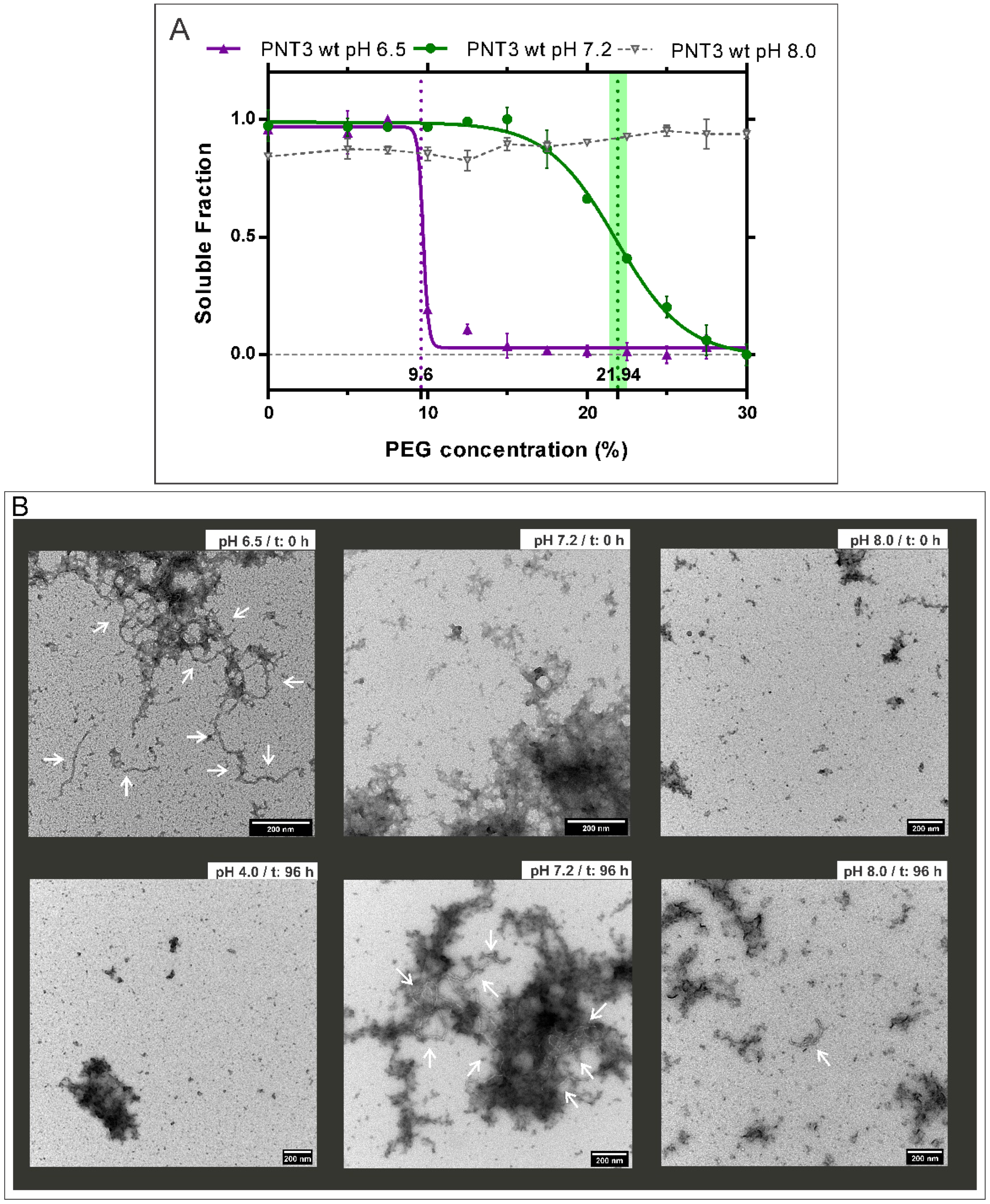
2.1. Influence of pH on the Formation of HeV PNT3 Amyloid-like Fibrils
2.2. Rational Design and Generation of PNT3 Variants
2.2.1. Design of PNT3 Variants Targeting the EYYY Motif
2.2.2. Design of HeV PNT3 Truncated Variants Devoid of the C-Terminal Region
2.2.3. Design of a HeV PNT3 Variant Bearing a Unique Cysteine
2.2.4. Expression and Purification of the PNT3 Variants
2.2.5. Conformational Characterization of the PNT3 Variants
2.3. Relevance of the PNT3 EYYY Motif in Fibrillation Abilities
2.3.1. Aggregation Propensity of the EYYY Motif PNT3 Variants
2.3.2. Congo Red Binding Abilities of PNT3 EYYY Motif Variants
2.3.3. Propensity and Time-Dependance of Fibrillation of the PNT3 EYYY Motif Variants Using Negative-Staining Transmission Electron Microscopy (ns-TEM)
2.4. Impact of the HeV PNT3 C-Terminal Region in Fibrillation Abilities
2.4.1. Aggregation Propensity of C-Terminally Truncated PNT3 Variants
2.4.2. CR Binding Ability of C-Terminally Truncated PNT3 Variants
2.4.3. Propensity and Kinetics of Fibrillation of C-Terminally Truncated PNT3 Variants Using ns-TEM Studies
2.5. Impact of a Cysteine in the HeV PNT3 Sequence on Fibrillation Abilities
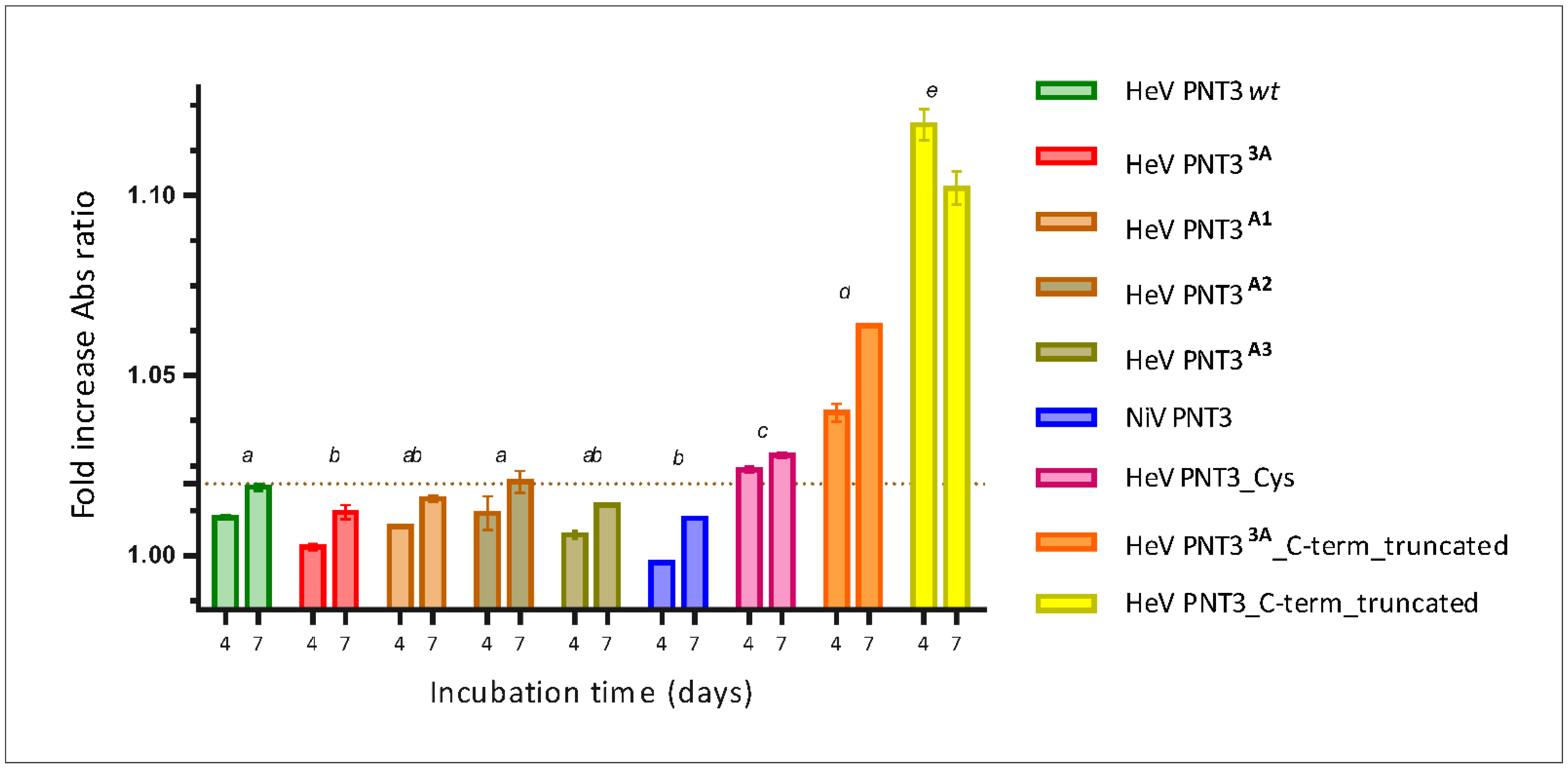
2.6. Characterization of the Aggregation Process by Taylor Dispersion Analysis
3. Materials and Methods
3.1. Generation of the Constructs
3.2. Proteins Expression and Purification
3.3. PEG Precipitation Assay (Relative Solubility)
3.4. Far-UV Circular Dichroism
3.5. Estimation of the Hydrodynamic Radius by SEC
3.6. Small-Angle X-ray Scattering (SAXS)
3.7. Congo Red Binding Assays
3.8. Negative-Staining Transmission Electron Microscopy (ns-TEM)
3.9. Kinetic Protein Aggregation Study by Taylor Dispersion Analysis (TDA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eaton, B.T.; Mackenzie, J.S.; Wang, L.F. Henipaviruses. In Fields Virology, 5th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 2007; pp. 1587–1600. [Google Scholar]
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef]
- Bloyet, L.M.; Welsch, J.; Enchery, F.; Mathieu, C.; de Breyne, S.; Horvat, B.; Grigorov, B.; Gerlier, D. HSP90 Chaperoning in Addition to Phosphoprotein Required for Folding but Not for Supporting Enzymatic Activities of Measles and Nipah Virus L Polymerases. J. Virol. 2016, 90, 6642–6656. [Google Scholar] [CrossRef] [Green Version]
- Abdella, R.; Aggarwal, M.; Okura, T.; Lamb, R.A.; He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl. Acad. Sci. USA 2020, 117, 4931–4941. [Google Scholar] [CrossRef]
- Bloyet, L.M.; Schramm, A.; Lazert, C.; Raynal, B.; Hologne, M.; Walker, O.; Longhi, S.; Gerlier, D. Regulation of measles virus gene expression by P protein coiled-coil properties. Sci. Adv. 2019, 5, eaaw3702. [Google Scholar] [CrossRef] [Green Version]
- Karlin, D.; Ferron, F.; Canard, B.; Longhi, S. Structural disorder and modular organization in Paramyxovirinae N and P. J. Gen. Virol. 2003, 84, 3239–3252. [Google Scholar] [CrossRef]
- Habchi, J.; Mamelli, L.; Darbon, H.; Longhi, S. Structural Disorder within Henipavirus Nucleoprotein and Phosphoprotein: From Predictions to Experimental Assessment. PLoS ONE 2010, 5, e11684. [Google Scholar] [CrossRef]
- Schiavina, M.; Salladini, E.; Murrali, M.G.; Tria, G.; Felli, I.C.; Pierattelli, R.; Longhi, S. Ensemble description of the intrinsically disordered N-terminal domain of the Nipah virus P/V protein from combined NMR and SAXS. Sci. Rep. 2020, 10, 19574. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.M.; Volchkov, V.; Blackledge, M.; et al. Structural Description of the Nipah Virus Phosphoprotein and Its Interaction with STAT1. Biophys. J. 2020, 118, 2470–2488. [Google Scholar] [CrossRef]
- Bruhn-Johannsen, J.F.; Barnett, K.; Bibby, J.; Thomas, J.; Keegan, R.; Rigden, D.; Bornholdt, Z.A.; Saphire, E.O. Crystal structure of the Nipah virus phosphoprotein tetramerization domain. J. Virol. 2014, 88, 758–762. [Google Scholar] [CrossRef] [Green Version]
- Blocquel, D.; Beltrandi, M.; Erales, J.; Barbier, P.; Longhi, S. Biochemical and structural studies of the oligomerization domain of the Nipah virus phosphoprotein: Evidence for an elongated coiled-coil homotrimer. Virology 2013, 446, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Beltrandi, M.; Blocquel, D.; Erales, J.; Barbier, P.; Cavalli, A.; Longhi, S. Insights into the coiled-coil organization of the Hendra virus phosphoprotein from combined biochemical and SAXS studies. Virology 2015, 477, 42–55. [Google Scholar] [CrossRef]
- Habchi, J.; Blangy, S.; Mamelli, L.; Ringkjobing Jensen, M.; Blackledge, M.; Darbon, H.; Oglesbee, M.; Shu, Y.; Longhi, S. Characterization of the interactions between the nucleoprotein and the phosphoprotein of Henipaviruses. J. Biol. Chem. 2011, 286, 13583–13602. [Google Scholar] [CrossRef] [Green Version]
- Communie, G.; Habchi, J.; Yabukarski, F.; Blocquel, D.; Schneider, R.; Tarbouriech, N.; Papageorgiou, N.; Ruigrok, R.W.; Jamin, M.; Ringkjøbing-Jensen, M.; et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 2013, 9, e1003631. [Google Scholar] [CrossRef]
- Douglas, J.; Drummond, A.J.; Kingston, R.L. Evolutionary history of cotranscriptional editing in the paramyxoviral phosphoprotein gene. Virus Evol. 2021, 7, veab028. [Google Scholar] [CrossRef]
- Salladini, E.; Delauzun, V.; Longhi, S. The Henipavirus V protein is a prevalently unfolded protein with a zinc-finger domain involved in binding to DDB1. Mol. Biosyst. 2017, 13, 2254–2267. [Google Scholar] [CrossRef]
- Pesce, G.; Gondelaud, F.; Ptchelkine, D.; Nilsson, J.F.; Bignon, C.; Cartalas, J.; Fourquet, P.; Longhi, S. Experimental Evidence of Intrinsic Disorder and Amyloid Formation by the Henipavirus W Proteins. Int. J. Mol. Sci. 2022, 23, 923. [Google Scholar] [CrossRef]
- Fontana, J.M.; Bankamp, B.; Rota, P.A. Inhibition of interferon induction and signaling by paramyxoviruses. Immunol. Rev. 2008, 225, 46–67. [Google Scholar] [CrossRef]
- Audsley, M.D.; Moseley, G.W. Paramyxovirus evasion of innate immunity: Diverse strategies for common targets. World J. Virol. 2013, 2, 57–70. [Google Scholar] [CrossRef]
- Tsimbalyuk, S.; Cross, E.M.; Hoad, M.; Donnelly, C.M.; Roby, J.A.; Forwood, J.K. The Intrinsically Disordered W Protein Is Multifunctional during Henipavirus Infection, Disrupting Host Signalling Pathways and Nuclear Import. Cells 2020, 9, 1913. [Google Scholar] [CrossRef]
- Salladini, E.; Gondelaud, F.; Nilsson, J.; Pesce, G.; Bignon, C.; Murrali, M.G.; Horvat, B.; Fabre, R.; Pierattelli, R.; Kajava, A.V.; et al. Identification of a region in the common amino-terminal domain of Hendra virus P, V and W proteins responsible for phase transition and amyloid formation. Biomolecules 2021, 11, 1324. [Google Scholar] [CrossRef]
- Gondelaud, F.; Pesce, G.; Nilsson, J.F.; Bignon, C.; Ptchelkine, D.; Gerlier, D.; Mathieu, C.; Longhi, S. Functional benefit of structural disorder for the replication of measles, Nipah and Hendra viruses. Essays Biochem. 2022, in press. [Google Scholar] [CrossRef]
- Kajava, A.V.; Baxa, U.; Steven, A.C. Beta arcades: Recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010, 24, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.B.; Znassi, N.; Château, M.T.; Kajava, A.V. A structure-based approach to predict predisposition to amyloidosis. Alzheimers Dement. 2015, 11, 681–690. [Google Scholar] [CrossRef]
- Swaminathan, R.; Ravi, V.K.; Kumar, S.; Kumar, M.V.; Chandra, N. Lysozyme: A model protein for amyloid research. Adv. Protein Chem. Struct. Biol. 2011, 84, 63–111. [Google Scholar] [CrossRef]
- Tseng, W.H.; Chen, S.H.; Hiramatsu, H. pH-controlled stacking direction of the β-strands in peptide fibrils. Sci. Rep. 2020, 10, 22199. [Google Scholar] [CrossRef]
- Pfefferkorn, C.M.; McGlinchey, R.P.; Lee, J.C. Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl. Acad. Sci. USA 2010, 107, 21447–21452. [Google Scholar] [CrossRef]
- Oeller, M.; Sormanni, P.; Vendruscolo, M. An open-source automated PEG precipitation assay to measure the relative solubility of proteins with low material requirement. Sci. Rep. 2021, 11, 21932. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Bignon, C.; Brocca, S.; Grandori, R.; Santambrogio, C.; Longhi, S. An arsenal of methods for the experimental characterization of intrinsically disordered proteins - How to choose and combine them? Arch. Biochem. Biophys. 2019, 676, 108055. [Google Scholar] [CrossRef]
- Anand, B.G.; Prajapati, K.P.; Shekhawat, D.S.; Kar, K. Tyrosine-Generated Nanostructures Initiate Amyloid Cross-Seeding in Proteins Leading to a Lethal Aggregation Trap. Biochemistry 2018, 57, 5202–5209. [Google Scholar] [CrossRef]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Gruet, A.; Dosnon, M.; Blocquel, D.; Brunel, J.; Gerlier, D.; Das, R.K.; Bonetti, D.; Gianni, S.; Fuxreiter, M.; Longhi, S.; et al. Fuzzy regions in an intrinsically disordered protein impair protein-protein interactions. FEBS J. 2016, 283, 576–594. [Google Scholar] [CrossRef] [Green Version]
- Troilo, F.; Bonetti, D.; Bignon, C.; Longhi, S.; Gianni, S. Understanding Intramolecular Crosstalk in an Intrinsically Disordered Protein. ACS Chem. Biol. 2019, 14, 337–341. [Google Scholar] [CrossRef]
- Deleanu, M.; Hernandez, J.F.; Cipelletti, L.; Biron, J.P.; Rossi, E.; Taverna, M.; Cottet, H.; Chamieh, J. Unraveling the Speciation of β-Amyloid Peptides during the Aggregation Process by Taylor Dispersion Analysis. Anal. Chem. 2021, 93, 6523–6533. [Google Scholar] [CrossRef]
- Deleanu, M.; Deschaume, O.; Cipelletti, L.; Hernandez, J.F.; Bartic, C.; Cottet, H.; Chamieh, J. Taylor Dispersion Analysis and Atomic Force Microscopy Provide a Quantitative Insight into the Aggregation Kinetics of Aβ (1-40)/Aβ (1-42) Amyloid Peptide Mixtures. ACS Chem. Neurosci. 2022, 13, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Deleanu, M.; Hernandez, J.F.; Cottet, H.; Chamieh, J. Taylor dispersion analysis discloses the impairment of Aβ peptide aggregation by the presence of a fluorescent tag. Electrophoresis 2022, in press.
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Brocca, S.; Testa, L.; Sobott, F.; Samalikova, M.; Natalello, A.; Papaleo, E.; Lotti, M.; De Gioia, L.; Doglia, S.M.; Alberghina, L.; et al. Compaction properties of an intrinsically disordered protein: Sic1 and its kinase-inhibitor domain. Biophys. J. 2011, 100, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Brookes, E.; Rocco, M. Recent advances in the UltraScan SOlution MOdeller (US-SOMO) hydrodynamic and small-angle scattering data analysis and simulation suite. Eur. Biophys. J. 2018, 47, 855–864. [Google Scholar] [CrossRef]
- Panjkovich, A.; Svergun, D.I. CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle X-ray scattering data. Bioinformatics 2018, 34, 1944–1946. [Google Scholar] [CrossRef] [Green Version]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Cryst. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Guinier, A. La diffraction des rayons X aux tres petits angles; application a l’etude de phenomenes ultramicroscopiques. Ann. Phys. 1939, 12, 161–237. [Google Scholar] [CrossRef]
- Guinier, A.; Fournet, F. Small Angle Scattering of X-rays; Wiley Interscience: New York, NY, USA, 1955. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameters in indirect-trasform methods using perceptual criteria. J. Appl. Cryst. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Bernado, P.; Blackledge, M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys. J. 2009, 97, 2839–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef] [PubMed]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015, 43, D357–D363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Mass | RSOBS | RSNF | RSPMG | RSU | RSIDP | RSOBS/RSNF | RSOBS/RSPMG | RSOBS/RSU | RSOBS/RSIDP | CI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HeV PNT3 wt | 15198 | 27.0 ± 0.3 | 19.4 | 27.9 | 33.9 | 30.0 | 1.39 | 0.97 | 0.80 | 0.90 | 0.47 ± 0.02 |
| HeV PNT33A | 14922 | 27.5 ± 0.4 | 19.3 | 27.7 | 33.5 | 30.0 | 1.42 | 0.99 | 0.82 | 0.92 | 0.42 ± 0.03 |
| HeV PNT3A1 | 15106 | 28.4 ± 0.4 | 19.4 | 27.9 | 33.8 | 30.0 | 1.36 | 0.94 | 0.78 | 0.88 | 0.37 ± 0.03 |
| HeV PNT3A2 | 15106 | 28.1 ± 0.3 | 19.4 | 27.9 | 33.8 | 30.0 | 1.31 | 0.91 | 0.76 | 0.85 | 0.39 ± 0.02 |
| HeV PNT3A3 | 15106 | 27.5 ± 0.2 | 19.4 | 27.9 | 33.8 | 30.0 | 1.42 | 0.99 | 0.81 | 0.92 | 0.44 ± 0.01 |
| NiV PNT3 | 14928 | 27.3 ± 0.7 | 19.3 | 27.7 | 33.5 | 30.5 | 1.41 | 0.99 | 0.82 | 0.90 | 0.44 ± 0.05 |
| HeV PNT3 C-term_truncated | 9020 | 19.0 ± 0.1 | 16.1 | 22.6 | 25.8 | 22.7 | 1.17 | 0.84 | 0.73 | 0.83 | 0.71 ± 0.01 |
| HeV PNT33A C-term_truncated | 8743 | 20.1 ± 0.6 | 16.0 | 22.4 | 25.4 | 22.7 | 1.26 | 0.90 | 0.79 | 0.89 | 0.56 ± 0.06 |
| HeV PNT3_Cys | 15230 | 28.0 ± 0.4 | 19.5 | 28.0 | 33.9 | 30.0 | 1.44 | 1.00 | 0.83 | 0.93 | 0.41 ± 0.03 |
| Proteins | I(0) cm−1 | Rg (Å) (Guinier) | Dmax (Å) | RgIDP (Å) | RgU (Å) |
|---|---|---|---|---|---|
| HeV PNT3 wt | 0.030 ± 1.8 × 10−4 | 36.67 ± 0.41 | 140 | 32.6 | 35.9 |
| HeV PNT33A | 0.022 ± 8.1 × 10−5 | 39.48 ± 0.33 | 144 | 32.6 | 35.9 |
| NiV PNT3 | 0.030 ± 8.2 × 10−5 | 37.37 ± 0.21 | 147 | 33.1 | 36.5 |
| HeV PNT3 C-term_truncated | 0.018 ± 4.1 × 10−5 | 27.53 ± 0.14 | 115 | 24.5 | 25.9 |
| HeV PNT33A C-term_truncated | 0.015 ± 5.6 × 10−5 | 27.34 ± 0.21 | 119 | 24.5 | 25.9 |
| Instrument | SOLEIL Synchrotron (Gif-sur-Yvette, France) Beamline Swing |
|---|---|
| X-rays wavelength (Å) | 1.033 |
| Energy (keV) | 12 |
| Detector type | Dectris EIGER 4M |
| Sample-to-detector distance (m) | 2.0 |
| q-range | 0.003 − 0.549 Å−1 |
| Temperature (°C) | 20 |
| Samples | |
| Concentration (mg mL−1) | 5 |
| Sample volume (µL) | 50 |
| Gel filtration column Flow rate (mL min−1) | AdvanceBio SEC 2.7 µm (Agilent) 0.3 |
| Buffer | 50 mM sodium phosphate pH 7.2 (buffer C) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsson, J.F.; Baroudi, H.; Gondelaud, F.; Pesce, G.; Bignon, C.; Ptchelkine, D.; Chamieh, J.; Cottet, H.; Kajava, A.V.; Longhi, S. Molecular Determinants of Fibrillation in a Viral Amyloidogenic Domain from Combined Biochemical and Biophysical Studies. Int. J. Mol. Sci. 2023, 24, 399. https://doi.org/10.3390/ijms24010399
Nilsson JF, Baroudi H, Gondelaud F, Pesce G, Bignon C, Ptchelkine D, Chamieh J, Cottet H, Kajava AV, Longhi S. Molecular Determinants of Fibrillation in a Viral Amyloidogenic Domain from Combined Biochemical and Biophysical Studies. International Journal of Molecular Sciences. 2023; 24(1):399. https://doi.org/10.3390/ijms24010399
Chicago/Turabian StyleNilsson, Juliet F., Hakima Baroudi, Frank Gondelaud, Giulia Pesce, Christophe Bignon, Denis Ptchelkine, Joseph Chamieh, Hervé Cottet, Andrey V. Kajava, and Sonia Longhi. 2023. "Molecular Determinants of Fibrillation in a Viral Amyloidogenic Domain from Combined Biochemical and Biophysical Studies" International Journal of Molecular Sciences 24, no. 1: 399. https://doi.org/10.3390/ijms24010399