

Investigation of the Fuzzy Complex between RSV Nucleoprotein and Phosphoprotein to Optimize an Inhibition Assay by Fluorescence Polarization
 , ,
, ,
Abstract
1. Introduction
2. Results
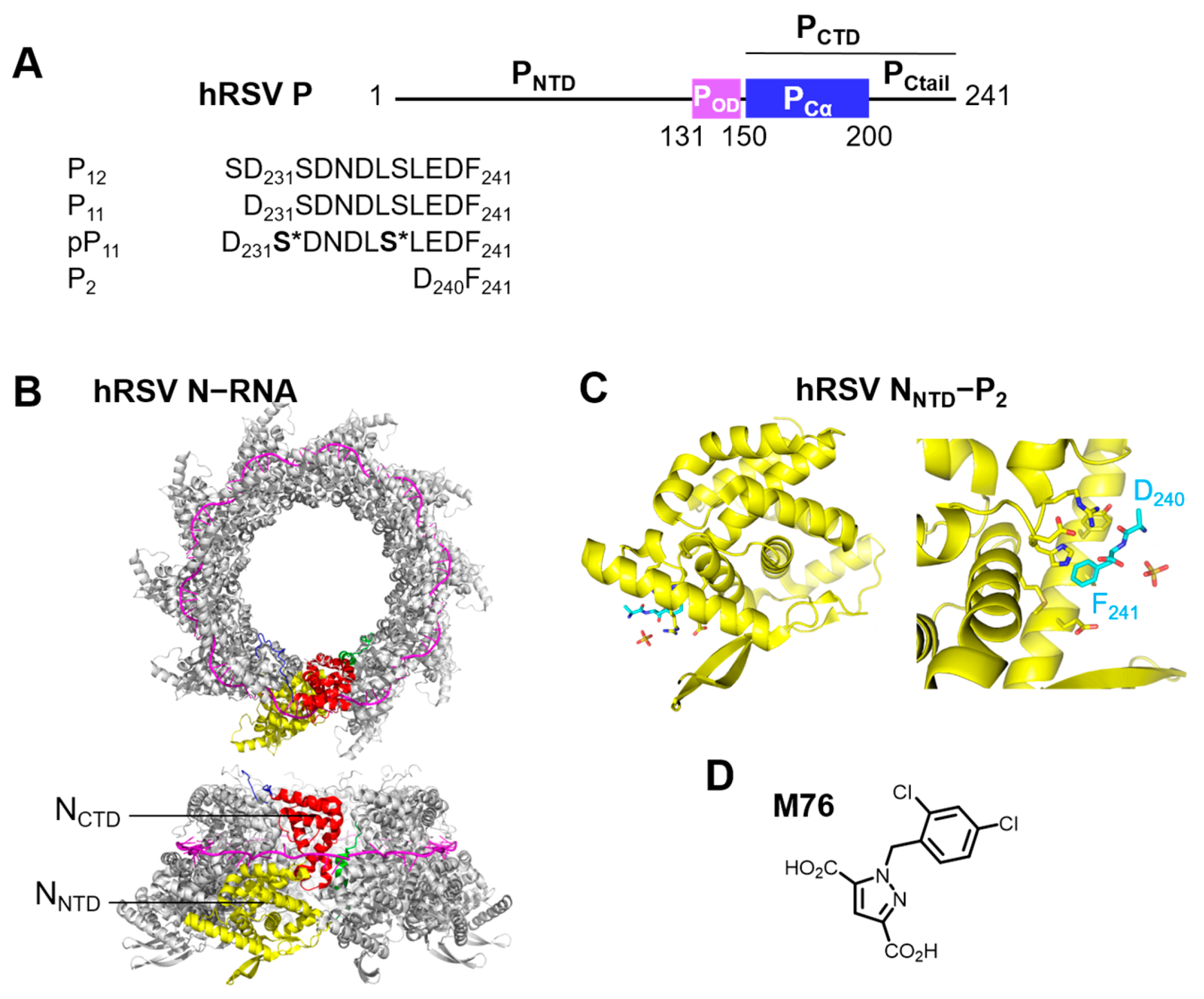
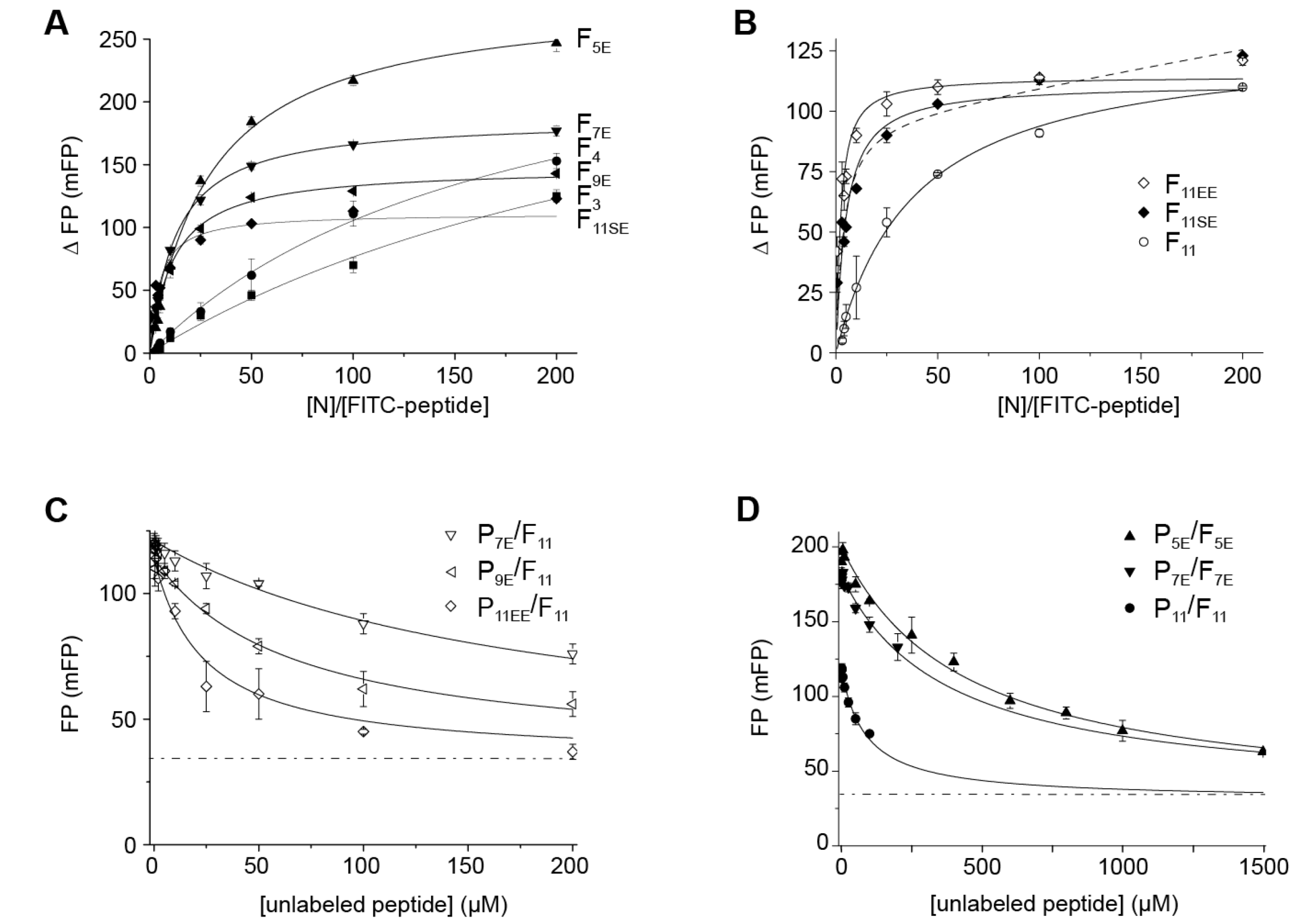
2.1. Phosphorylation of an RSV PCtail-Derived Peptide Increases Affinity for RSV NNTD
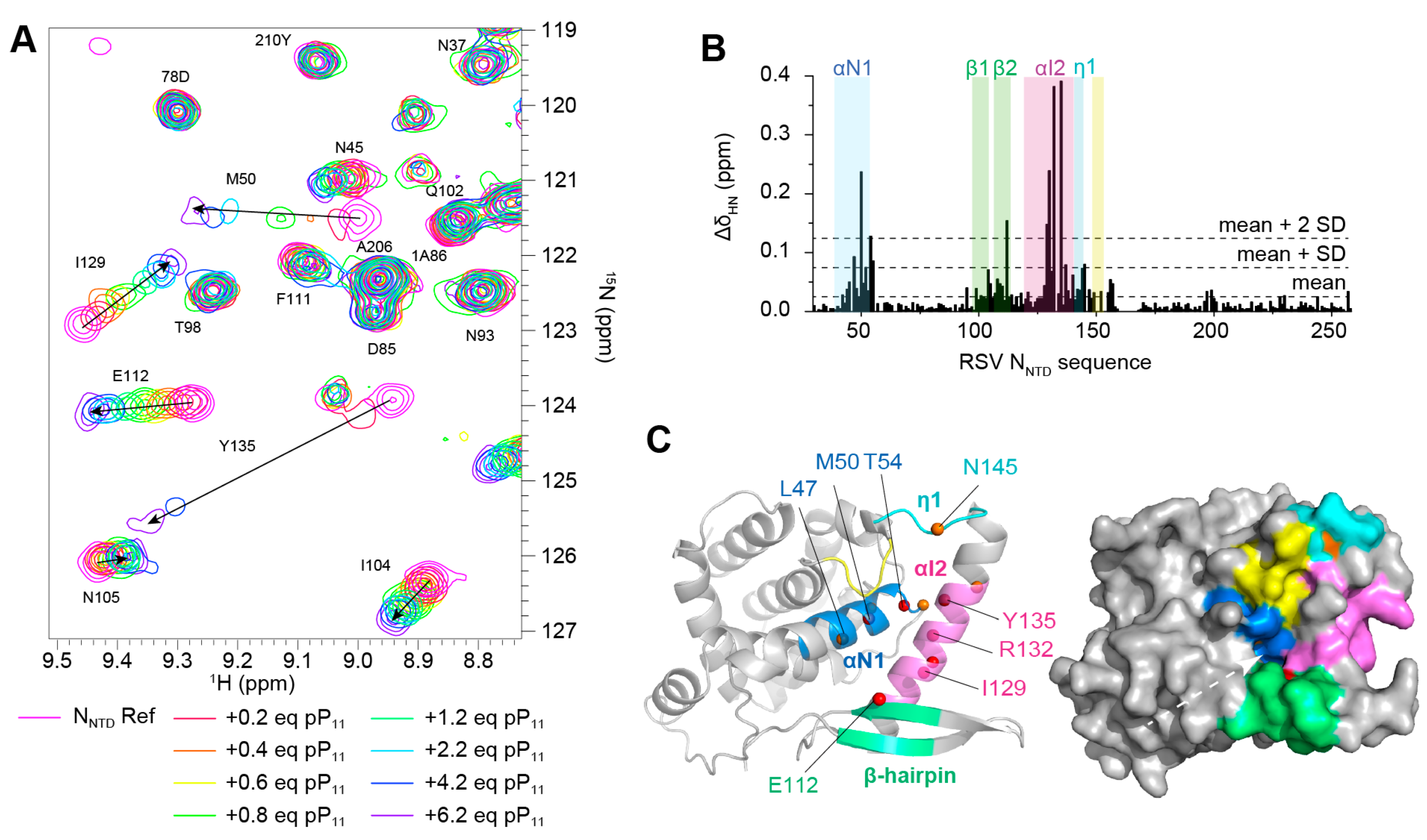
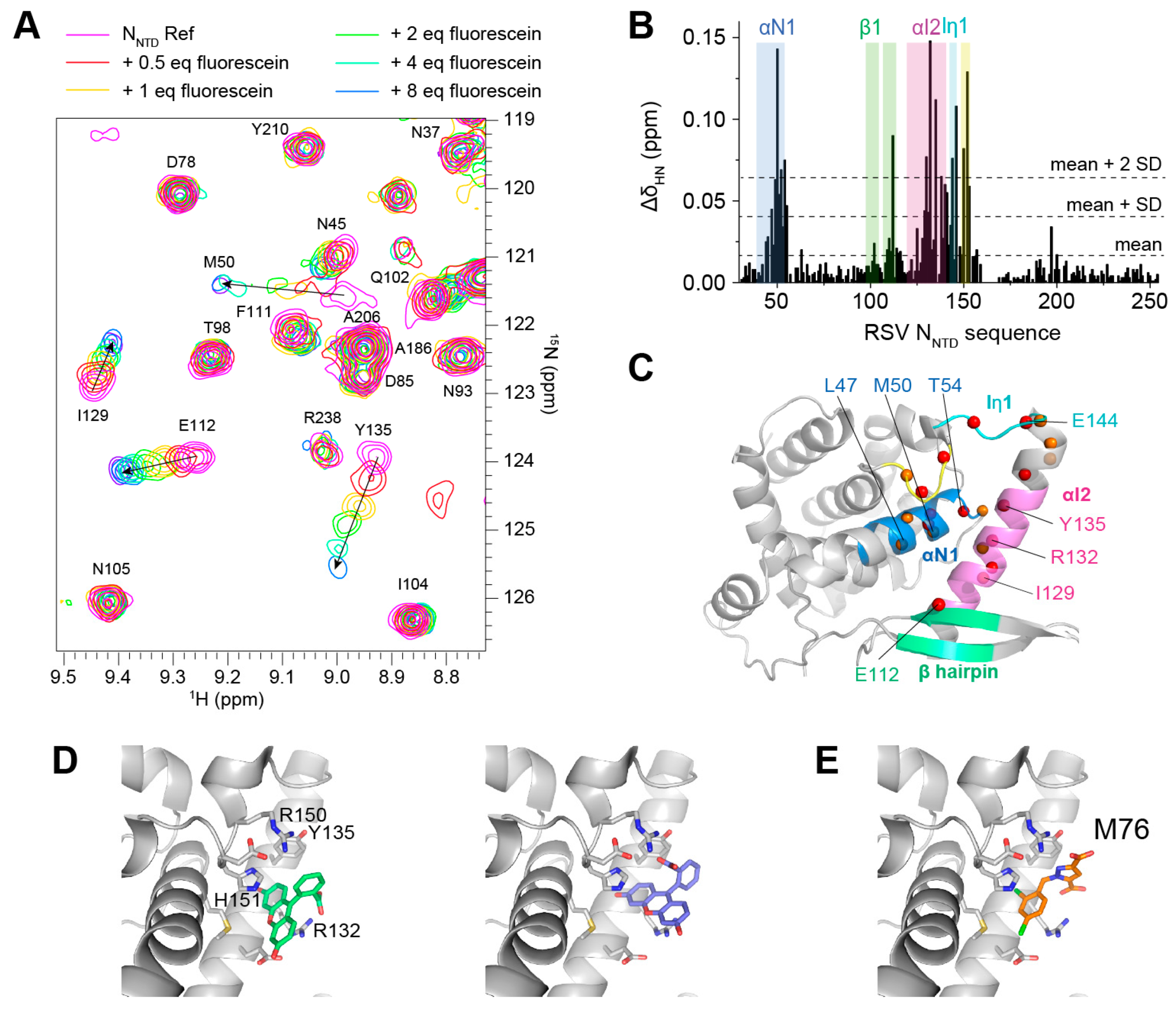
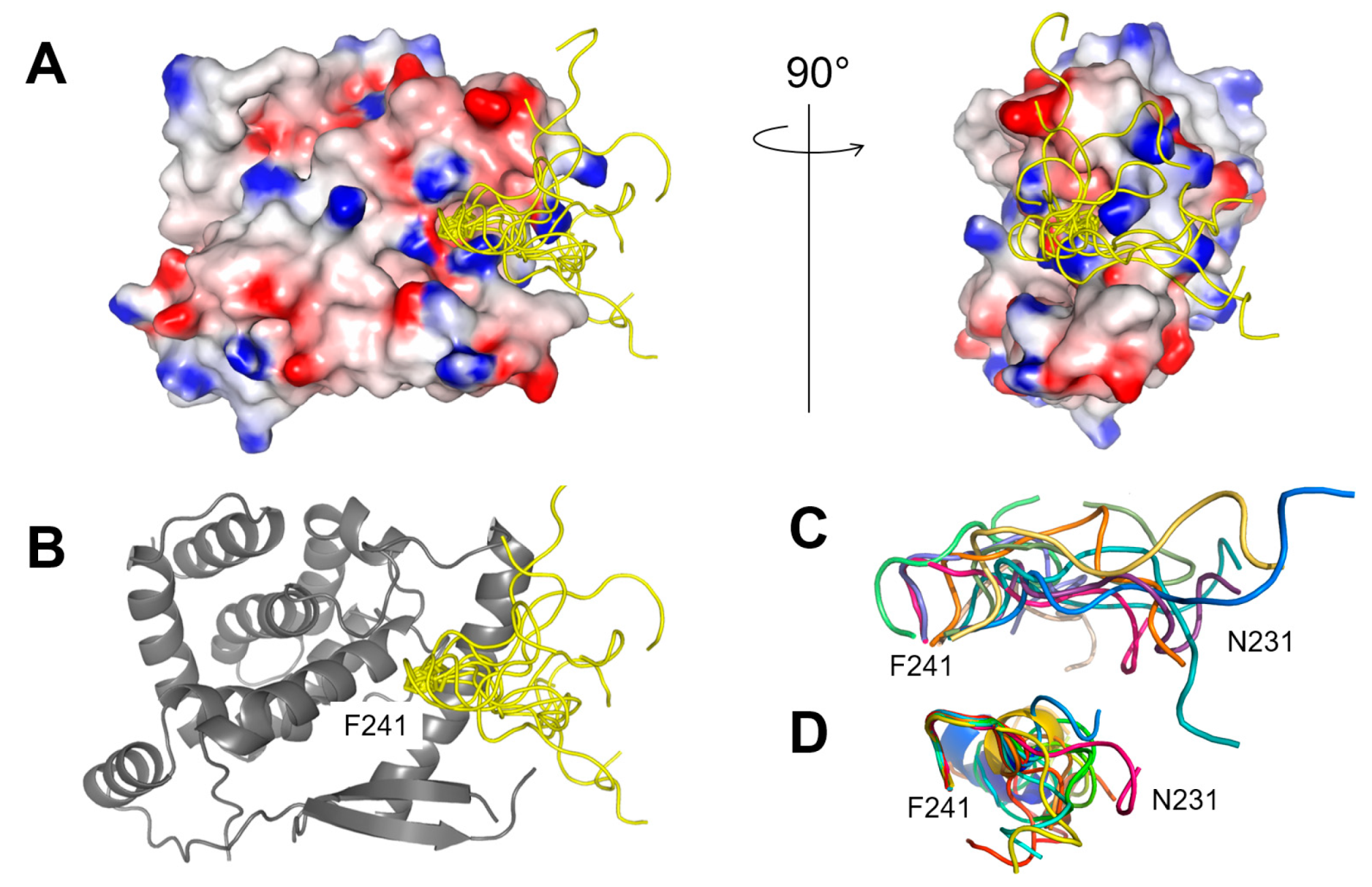
2.2. Fluorescence Polarization Reveals a Potential Secondary Binding Site on RSV NNTD for Fluorescein-Labeled RSV P11 Peptide
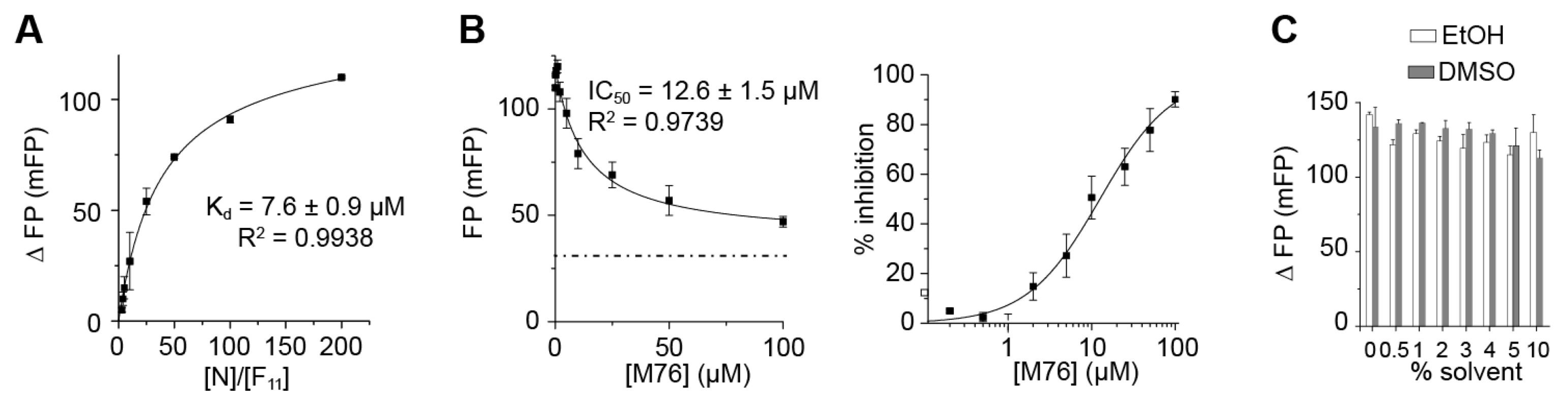
2.3. The Complex betweeen Fluorescein-Labeled C-Terminal P Peptides and Full-Length N Protein Provides a Robust Model of the RSV N-P Interaction for FP Measurements
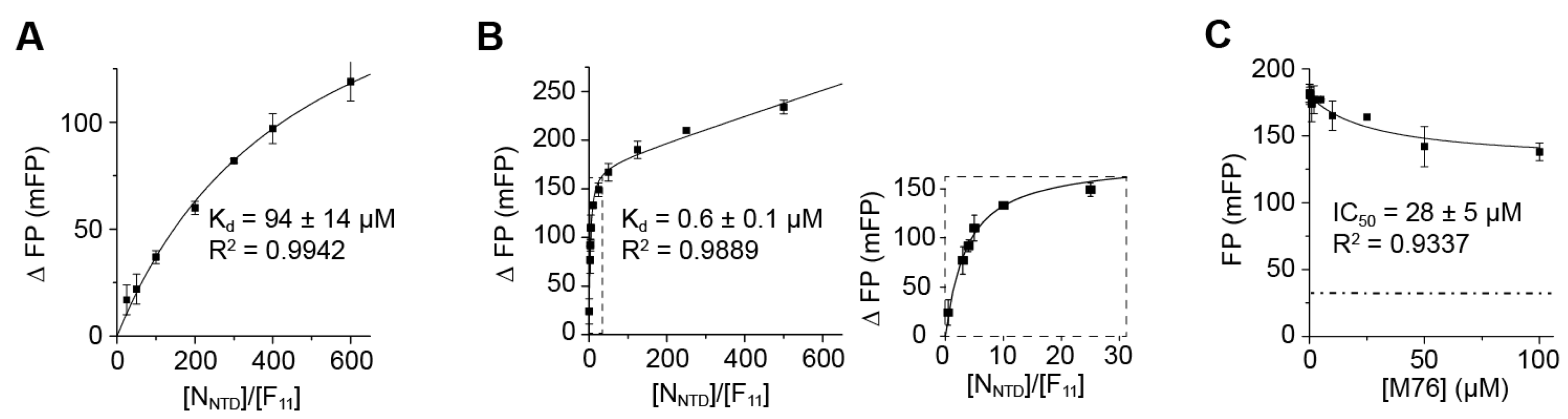
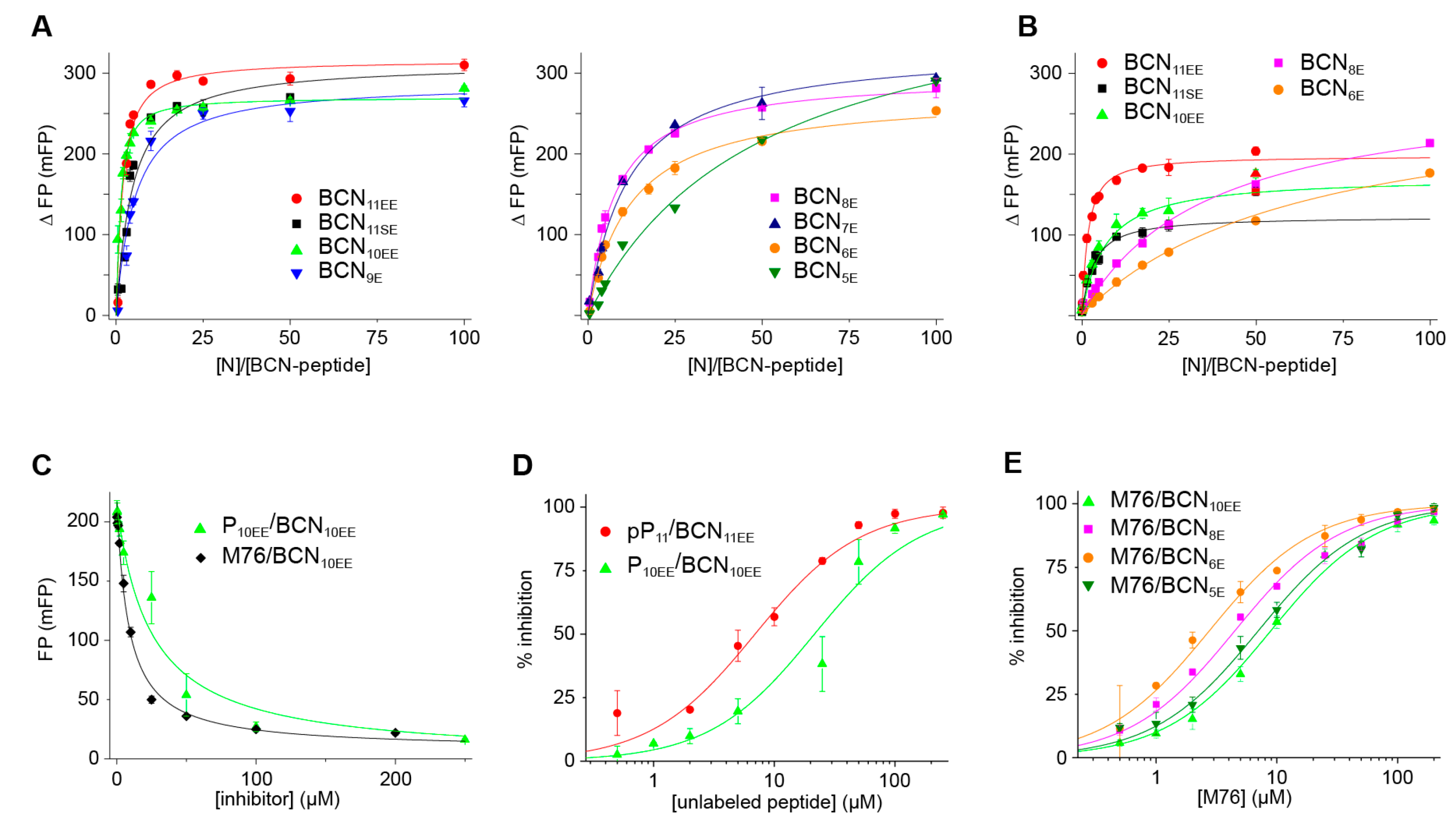
2.4. Measurement of the Binding Affinity of RSV Fluorescein-P Peptides for RSV N-RNA and Influence of Peptide Length on Fluorescent Label Mobility
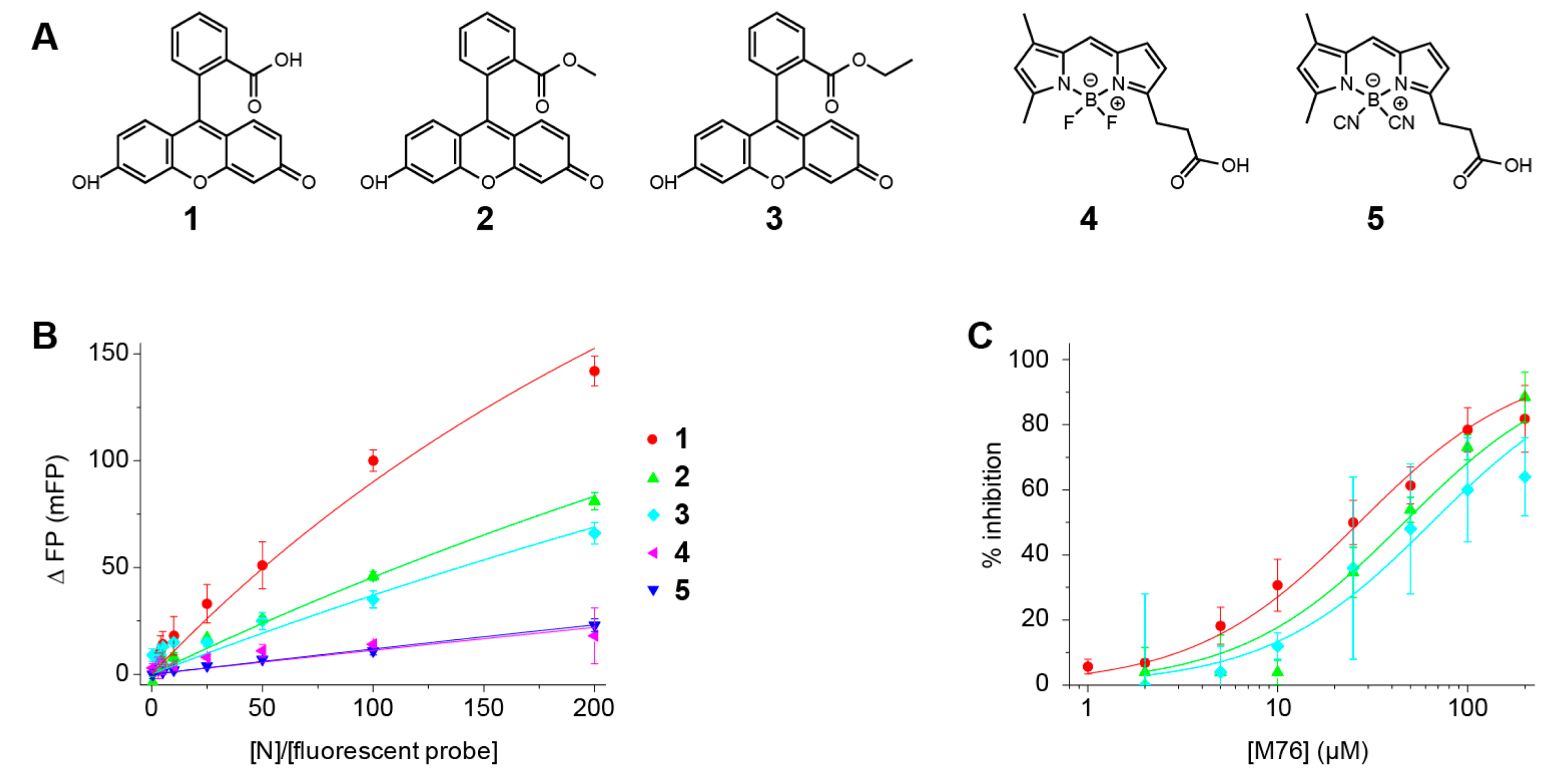
2.5. Fluorescein Binds to the RSV P-Binding Site on RSV N Protein
2.6. An FP Assay for the RSV N-P Interaction Using Full-Length N and BODIPY FL-Labeled P Peptides
3. Discussion
4. Materials and Methods
4.1. Materials
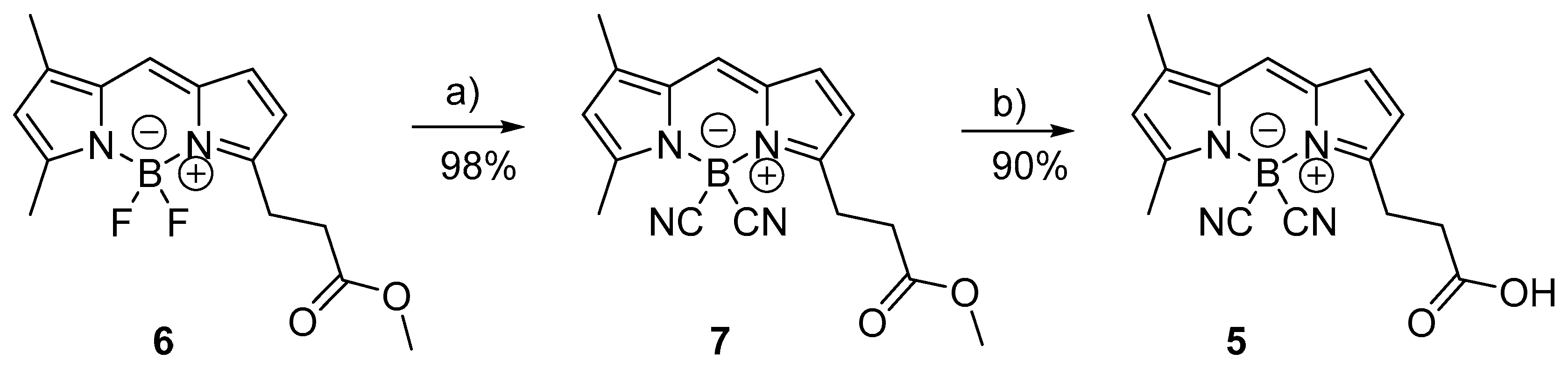
4.2. Synthesis of Fluorescent Molecules
4.3. Peptide Synthesis
4.3.1. Procedure A: Fmoc Removal
4.3.2. Procedure B: Coupling Steps
4.3.3. Procedure C: Coupling of Dicyano BODIPY FL
4.3.4. Procedure D: Resin Cleavage and Protecting Groups Removal
4.4. Peptide Purification and Yields
4.4.1. Unlabeled Peptides
- P3 peptide was obtained as a white powder (34.0 mg, 50%). Analytical UPLC tr = 0.87 min; ESI-MS (positive mode) calculated for [C18H23N3O8], 409.2; found m/z, 410.4 (M + H)+. [Focused gradient 5–25% of B in 15 min].
- P4 peptide was obtained as a white powder (49.0 mg, 59%). Analytical UPLC tr = 1.01 min; ESI-MS (positive mode) calculated for [C24H34N4O9], 522.2; found m/z, 523.5 (M + H)+. [Focused gradient 10–30% of B in 15 min].
- P5E peptide was obtained as a white powder (59.0 mg, 59%). Analytical UPLC tr = 1.00 min; ESI-MS (positive mode) calculated for [C29H41N5O12], 651.3; found m/z, 652.6 (M + H)+. [Focused gradient 15–35% of B in 15 min].
- P7E peptide was obtained as a white powder (43.0 mg, 33%). Analytical UPLC tr = 1.43 min; ESI-MS (positive mode) calculated for [C39H57N7O16], 879.4; found m/z, 880.8 (M + H)+. [Focused gradient 25–35% of B in 15 min].
- P9E peptide was obtained as a white powder (27.0 mg, 17%). Analytical UPLC tr = 1.29 min; ESI-MS (positive mode) calculated for [C47H68N10O21], 1108.5; found m/z, 1109.7 (M + H)+. [Focused gradient 20–30% of B in 15 min].
- P11EE: SPPS of P11EE peptide was performed on 150 mg of 2-CTC resin. The peptide P11EE was obtained as a white powder (28.0 mg, 20%). Analytical UPLC tr = 1.28 min; ESI-MS (positive mode) calculated for [C56H80N12O27], 1352.5; found m/z, 1353.9 (M + H)+. [Focused gradient 20–30% of B in 15 min].
- P11SE: SPPS of P11SE peptide was performed on 15.0 mg of 2-CTC resin. P11SE was obtained as a white powder (4.50 mg) and was used without further purification for F11SE synthesis.
4.4.2. Dicyano BODIPY FL-Labeled BCNn Peptides
- BCN5E: Starting from P5E on resin (8.71 µmol), BCN5E was obtained as a red powder (2.63 mg, 32%). Analytical UPLC tr = 1.61 min; ESI-MS (negative mode) calculated for [C45H54BN9O13], 939.8; found m/z, 939.4 (M − H)−. [Focused gradient 38–48% of B in 15 min].
- BCN6E: Starting from P6E on resin (8.71 µmol), BCN6E was obtained as a red powder (1.60 mg, 18%). Analytical UPLC tr = 1.74 min; ESI-MS (negative mode) calculated for [C51H65BN10O14], 1052.5; found m/z, 1051.2 (M − H)−. [Focused gradient 43–53% of B in 15 min].
- BCN7E: Starting from P7E on resin (8.71 µmol), BCN7E was obtained as a red powder (2.22 mg, 22%). Analytical UPLC tr = 1.66 min; ESI-MS (negative mode) calculated for [C55H70BN11O17], 1167.5; found m/z, 1166.9 (M − H)−. [Focused gradient 40–50% of B in 15 min].
- BCN8E: Starting from P8E on resin (8.71 µmol), BCN8E was obtained as a red powder (2.00 mg, 18%). Analytical UPLC tr = 1.56 min; ESI-MS (positive mode) calculated for [C59H76BN13O19], 1282.6; found m/z, 1283.9 (M + H)+. [Focused gradient 36–46% of B in 15 min].
- BCN9E: Starting from P9E on resin (8.71 µmol), B9E was obtained as a red powder (1.17 mg, 10%). Analytical UPLC tr = 1.53 min; ESI-MS (positive mode) calculated for [C63H81BN14O22], 1396.6; found m/z, 1398.2 (M + H)+. [Focused gradient 35–45% of B in 15 min].
- BCN10EE: Starting from P10EE on resin (8.71 µmol), BCN10EE was obtained as a red powder (1.00 mg, 8%). Analytical UPLC tr = 1.50 min; ESI-MS (negative mode) calculated for [C68H88BN15O25], 1525.6; found m/z, 1525,1(M − H)−. [Focused gradient 34–44% of B in 15 min]. UV-Visible absorption spectrum and fluorescence excitation/emission spectra are given in Figure S33.
- BCN11EE: Starting from P11EE on resin (8.71 µmol), BCN11EE was obtained as a red powder (1.01 mg, 11%). Analytical UPLC tr = 1.49 min; ESI-MS (negative mode) calculated for [C72H93BN16O28], 1640.6; found m/z, 1640.3 (M − H)−. [Focused gradient 33–43% of B in 15 min].
- BCN11SE: Starting from P11SE on resin (4.35 µmol), BCN11SE was obtained as a red powder (1.12 mg, 16%). Analytical UPLC tr = 1.48 min; ESI-MS (negative mode) calculated for [C70H91BN16O27], 1598.6; found m/z, 1598.2 (M − H)−. [Focused gradient 32–42% of B in 15 min].
4.4.3. Fluorescein-Labeled Fn-Peptides
- F3: Starting from P3 (5.00 mg, 9.55 µmol), F3 was obtained as a yellow powder (3.00 mg, 48%). Analytical UPLC tr = 1.31 min; ESI-MS (negative mode) calculated for [C39H34N4O13S], 798.2; found m/z, 797.6 (M − H)−. [Focused gradient 25–45% of B in 15 min].
- F4: Starting from P4 (5.00 mg, 7.85 µmol), F4 was obtained as a yellow powder (4.30 mg, 62%). Analytical UPLC tr = 2.12 min; ESI-MS (negative mode) calculated for [C45H45N5O14S], 911.3; found m/z, 910.6 (M − H)−. [Focused gradient 55–65% of solvent D consisted of MeOH containing 9.9% (v/v) H2O and 0.1% TFA in 15 min].
- F5E: Starting from P5E (5.00 mg, 6.53 µmol), F5E was obtained as a yellow powder (3.60 mg, 53%). Analytical UPLC tr = 0.98 min; ESI-MS (positive mode) calculated for [C50H52N6O17S], 1040.3; found m/z, 1041.8 (M + H)+. [Focused gradient 35–45% of B in 15 min].
- F7E: Starting from P7E (5.00 mg, 5.03 µmol), F7E was obtained as a yellow powder (2.60 mg, 50%). Analytical UPLC tr = 1.53 min; ESI-MS (positive mode) calculated for [C60H68N8O21S], 1268.4; found m/z, 1270.0 (M + H)+. [Focused gradient 30–50% of B in 15 min].
- F9E: Starting from P9E (5.00 mg, 4.09 µmol), F9E was obtained as a yellow powder (1.40 mg, 22%) Analytical UPLC tr = 1.42 min; ESI-MS (positive mode) calculated for [C68H79N11O26S], 1497.5; found m/z, 1499.7 (M + H)+. [Focused gradient 30–50% of B in 15 min].
- F11: Starting from commercial P11 (4.30 mg, 3.11 µmol), F11SS was obtained as a yellow powder (3.5 mg, 68%). Analytical UPLC tr = 1.45 min; ESI-MS (positive mode) calculated for [C73H87N13O30S], 1657.5; found m/z, 1659.8 (M + H)+. [Focused gradient 25–45% of B in 15 min].
- F11SE: Starting from P11SE (4.50 mg, 3.43 µmol), F11SE was obtained as a yellow powder (0.90 mg, 15%). Analytical UPLC tr = 1.39 min; ESI-MS (negative mode) calculated for [C75H89N13O31S], 1699.6; found m/z, 1697.7 (M − H)−. [Focused gradient 25–45% of B in 15 min].
- F11EE: Starting from P11EE (5 mg, 3.41 µmol), F11EE was obtained as a yellow powder (2.40 mg, 40%). Analytical UPLC tr = 1.39 min; ESI-MS (positive mode) calculated for [C77H91N13O32S], 1741.6; found m/z, 1742.6 (M + H)+. [Focused gradient 25–45% of B in 15 min].
- pF11: Starting from commercial pP11 (4.20 mg, 2.93 µmol), pF11 was obtained as a yellow powder (1.10 mg, 20%). Analytical UPLC tr = 1.36 min; ESI-MS (negative mode) calculated for [C73H89N13O36P2S], 1817.5; found m/z, 908.7 (M − 2H)2−. [Focused gradient 25–45% of B in 15 min].
4.5. Bacterial Expression and Purification of RSV N Protein
4.6. NMR Measurements
4.7. Fluorescence Polarization Measurements
4.8. Complex Modelling with Haddock
4.9. Docking of Small Compounds
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Brien, K.L.; Baggett, H.C.; Brooks, W.A.; Feikin, D.R.; Hammitt, L.L.; Higdon, M.M.; Howie, S.R.; Knoll, M.D.; Kotloff, K.L.; Levine, O.S.; et al. Causes of severe pneumonia requiring hospital admission in children without HIV infection from Africa and Asia: The PERCH multi-country case-control study. Lancet 2019, 394, 757–779. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Coultas, J.A.; Smyth, R.; Openshaw, P.J. Respiratory syncytial virus (RSV): A scourge from infancy to old age. Thorax 2019, 74, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Cockerill, G.S.; Good, J.A.D.; Mathews, N. State of the Art in Respiratory Syncytial Virus Drug Discovery and Development. J. Med. Chem. 2019, 62, 3206–3227. [Google Scholar] [CrossRef]
- Elawar, F.; Oraby, A.K.; Kieser, Q.; Jensen, L.D.; Culp, T.; West, F.G.; Marchant, D.J. Pharmacological targets and emerging treatments for respiratory syncytial virus bronchiolitis. Pharmacol. Ther. 2021, 220, 107712. [Google Scholar] [CrossRef]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, Detection, and New Options for Prevention and Treatment. Clin. Microbiol. Rev. 2017, 30, 277–319. [Google Scholar] [CrossRef]
- Hammitt, L.L.; Dagan, R.; Yuan, Y.; Baca Cots, M.; Bosheva, M.; Madhi, S.A.; Muller, W.J.; Zar, H.J.; Brooks, D.; Grenham, A.; et al. Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N. Engl. J. Med. 2022, 386, 837–846. [Google Scholar] [CrossRef]
- Thornhill, E.M.; Salpor, J.; Verhoeven, D. Respiratory syntycial virus: Current treatment strategies and vaccine approaches. Antivir. Chem. Chemother. 2020, 28, 2040206620947303. [Google Scholar] [CrossRef]
- Mazur, N.I.; Terstappen, J.; Baral, R.; Bardají, A.; Beutels, P.; Buchholz, U.J.; Cohen, C.; Crowe, J.E.; Cutland, C.L.; Eckert, L.; et al. Respiratory syncytial virus prevention within reach: The vaccine and monoclonal antibody landscape. Lancet Infect. Dis. 2022, 23, e2–e21. [Google Scholar] [CrossRef]
- Heylen, E.; Neyts, J.; Jochmans, D. Drug candidates and model systems in respiratory syncytial virus antiviral drug discovery. Biochem. Pharmacol. 2017, 127, 1–12. [Google Scholar] [CrossRef]
- Nicholson, E.G.; Munoz, F.M. A Review of Therapeutics in Clinical Development for Respiratory Syncytial Virus and Influenza in Children. Clin. Ther. 2018, 40, 1268–1281. [Google Scholar] [CrossRef]
- Stevens, M.; Rusch, S.; DeVincenzo, J.; Kim, Y.-I.; Harrison, L.; Meals, E.A.; Boyers, A.; Fok-Seang, J.; Huntjens, D.; Lounis, N.; et al. Antiviral Activity of Oral JNJ-53718678 in Healthy Adult Volunteers Challenged with Respiratory Syncytial Virus: A Placebo-Controlled Study. J. Infect. Dis. 2018, 218, 748–756. [Google Scholar] [CrossRef]
- DeVincenzo, J.P.; Whitley, R.J.; Mackman, R.L.; Scaglioni-Weinlich, C.; Harrison, L.; Farrell, E.; McBride, S.; Lambkin-Williams, R.; Jordan, R.; Xin, Y.; et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2014, 371, 711–722. [Google Scholar] [CrossRef]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and Characterization of ALX-0171, a Potent Novel Therapeutic Nanobody for the Treatment of Respiratory Syncytial Virus Infection. Antimicrob. Agents Chemother. 2016, 60, 6–13. [Google Scholar] [CrossRef]
- DeVincenzo, J.P.; McClure, M.W.; Symons, J.A.; Fathi, H.; Westland, C.; Chanda, S.; Lambkin-Williams, R.; Smith, P.; Zhang, Q.; Beigelman, L.; et al. Activity of Oral ALS-008176 in a Respiratory Syncytial Virus Challenge Study. N. Engl. J. Med. 2015, 373, 2048–2058. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; Ayllon, M.A.; Bao, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef]
- Collins, P.L.; Karron, R.A. Respiratory Syncytial Virus and Metapneumovirus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippinscot Williams & Wilkins, Wolters Kluwer: Philadelphia, PA, USA, 2013; pp. 1086–1123. [Google Scholar]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.E.; Duquerroy, S.; Galloux, M.; Loney, C.; Conner, E.; Eleouet, J.F.; Rey, F.A.; Bhella, D. The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J. Gen. Virol. 2013, 94, 1734–1738. [Google Scholar] [CrossRef]
- Galloux, M.; Tarus, B.; Blazevic, I.; Fix, J.; Duquerroy, S.; Eleouet, J.F. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J. Virol. 2012, 86, 8375–8387. [Google Scholar] [CrossRef]
- Fearns, R. The Respiratory Syncytial Virus Polymerase: A Multitasking Machine. Trends Microbiol. 2019, 27, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Gilman, M.S.A.; Liu, C.; Fung, A.; Behera, I.; Jordan, P.; Rigaux, P.; Ysebaert, N.; Tcherniuk, S.; Sourimant, J.; Eleouet, J.F.; et al. Structure of the Respiratory Syncytial Virus Polymerase Complex. Cell 2019, 179, 193–204 e114. [Google Scholar] [CrossRef] [PubMed]
- Galloux, M.; Risso-Ballester, J.; Richard, C.A.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Minimal Elements Required for the Formation of Respiratory Syncytial Virus Cytoplasmic Inclusion Bodies In Vivo and In Vitro. MBio 2020, 11, e01202-20. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.; Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentín, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing virus condensation. PLoS Pathog. 2021, 17, e1009926. [Google Scholar] [CrossRef]
- Pereira, N.; Cardone, C.; Lassoued, S.; Galloux, M.; Fix, J.; Assrir, N.; Lescop, E.; Bontems, F.; Eleouet, J.F.; Sizun, C. New Insights into Structural Disorder in Human Respiratory Syncytial Virus Phosphoprotein and Implications for Binding of Protein Partners. J. Biol. Chem. 2017, 292, 2120–2131. [Google Scholar] [CrossRef]
- Cardone, C.; Caseau, C.-M.; Bardiaux, B.; Thureaux, A.; Galloux, M.; Bajorek, M.; Eléouët, J.-F.; Litaudon, M.; Bontems, F.; Sizun, C. A Structural and Dynamic Analysis of the Partially Disordered Polymerase-Binding Domain in RSV Phosphoprotein. Biomolecules 2021, 11, 1225. [Google Scholar] [CrossRef]
- Noval, M.G.; Esperante, S.A.; Molina, I.G.; Chemes, L.B.; Prat-Gay, G. Intrinsic disorder to order transitions in the scaffold phosphoprotein P from the respiratory syncytial virus RNA-polymerase complex. Biochemistry 2016, 55, 1441–1454. [Google Scholar] [CrossRef]
- Cardone, C.; Caseau, C.-M.; Pereira, N.; Sizun, C. Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility. Int. J. Mol. Sci. 2021, 22, 1537. [Google Scholar] [CrossRef]
- Galloux, M.; Gabiane, G.; Sourimant, J.; Richard, C.A.; England, P.; Moudjou, M.; Aumont-Nicaise, M.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Identification and characterization of the binding site of the respiratory syncytial virus phosphoprotein to RNA-free nucleoprotein. J. Virol. 2015, 89, 3484–3496. [Google Scholar] [CrossRef]
- Tran, T.L.; Castagne, N.; Bhella, D.; Varela, P.F.; Bernard, J.; Chilmonczyk, S.; Berkenkamp, S.; Benhamo, V.; Grznarova, K.; Grosclaude, J.; et al. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J. Gen. Virol. 2007, 88, 196–206. [Google Scholar] [CrossRef]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagne, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef]
- Ouizougun-Oubari, M.; Pereira, N.; Tarus, B.; Galloux, M.; Lassoued, S.; Fix, J.; Tortorici, M.A.; Hoos, S.; Baron, B.; England, P.; et al. A Druggable Pocket at the Nucleocapsid/Phosphoprotein Interaction Site of Human Respiratory Syncytial Virus. J. Virol. 2015, 89, 11129–11143. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef]
- Sharma, R.; Raduly, Z.; Miskei, M.; Fuxreiter, M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015, 589, 2533–2542. [Google Scholar] [CrossRef]
- Mazumder, B.; Barik, S. Requirement of casein kinase II-mediated phosphorylation for the transcriptional activity of human respiratory syncytial viral phosphoprotein P: Transdominant negative phenotype of phosphorylation-defective P mutants. Virology 1994, 205, 104–111. [Google Scholar] [CrossRef]
- Sanchez-Seco, M.P.; Navarro, J.; Martinez, R.; Villanueva, N. C-terminal phosphorylation of human respiratory syncytial virus P protein occurs mainly at serine residue 232. J. Gen. Virol. 1995, 76 Pt. 2, 425–430. [Google Scholar] [CrossRef]
- Villanueva, N.; Hardy, R.; Asenjo, A.; Yu, Q.; Wertz, G. The bulk of the phosphorylation of human respiratory syncytial virus phosphoprotein is not essential but modulates viral RNA transcription and replication. J. Gen. Virol. 2000, 81, 129–133. [Google Scholar] [CrossRef]
- Lu, B.; Ma, C.H.; Brazas, R.; Jin, H. The major phosphorylation sites of the respiratory syncytial virus phosphoprotein are dispensable for virus replication in vitro. J. Virol. 2002, 76, 10776–10784. [Google Scholar] [CrossRef]
- Kaul, T.N.; Middleton, E.; Ogra, P.L. Antiviral effect of flavonoids on human viruses. J. Med. Virol. 1985, 15, 71–79. [Google Scholar] [CrossRef]
- Sa, J.M.; Piloto, J.V.; Cilli, E.M.; Tasic, L.; Fossey, M.A.; Almeida, F.C.L.; Souza, F.P.; Caruso, I.P. Hesperetin targets the hydrophobic pocket of the nucleoprotein/phosphoprotein binding site of human respiratory syncytial virus. J. Biomol. Struct. Dyn. 2020, 40, 2156–2168. [Google Scholar] [CrossRef]
- Rhodin, M.H.J.; McAllister, N.V.; Castillo, J.; Noton, S.L.; Fearns, R.; Kim, I.J.; Yu, J.; Blaisdell, T.P.; Panarese, J.; Shook, B.C.; et al. EDP-938, a novel nucleoprotein inhibitor of respiratory syncytial virus, demonstrates potent antiviral activities in vitro and in a non-human primate model. PLoS Pathog. 2021, 17, e1009428. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Fluorescence Anisotropy. In Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publisher: New York, NY, USA, 1999; pp. 291–319. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, Q.; Berezin, M.Y. Fluorescence anisotropy (polarization): From drug screening to precision medicine. Expert Opin. Drug Discov. 2015, 10, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Gao, N.; O’Connell, N.; Hu, J.; Thresher, J.; Gu, R.F.; Overman, R.; Hardern, I.M.; Sproat, G.G. Quantitative investigation of the affinity of human respiratory syncytial virus phosphoprotein C-terminus binding to nucleocapsid protein. Virol. J. 2014, 11, 191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, Y.; Jiang, Y.; Ben-Amotz, D. Detection of amino acid and peptide phosphate protonation using Raman spectroscopy. Anal. Biochem. 2005, 343, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Sportsman, J.R. Fluorescence anisotropy in pharmacologic screening. Methods Enzymol. 2003, 361, 505–529. [Google Scholar] [CrossRef]
- Hall, M.D.; Yasgar, A.; Peryea, T.; Braisted, J.C.; Jadhav, A.; Simeonov, A.; Coussens, N.P. Fluorescence polarization assays in high-throughput screening and drug discovery: A review. Methods Appl. Fluoresc. 2016, 4, 022001. [Google Scholar] [CrossRef]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef]
- Martin, M.M.; Lindqvist, L. The pH dependence of fluorescein fluorescence. J. Lumin. 1975, 10, 381–390. [Google Scholar] [CrossRef]
- Klonis, N.; Sawyer, W.H. Spectral properties of the prototropic forms of fluorescein in aqueous solution. J. Fluoresc. 1996, 6, 147–157. [Google Scholar] [CrossRef]
- Le Guern, F.; Mussard, V.; Gaucher, A.; Rottman, M.; Prim, D. Fluorescein Derivatives as Fluorescent Probes for pH Monitoring along Recent Biological Applications. Int. J. Mol. Sci. 2020, 21, 9217. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2022. [Google Scholar]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef]
- Uriel, C.; Gómez, A.M.; García Martínez de la Hidalga, E.; Bañuelos, J.; Garcia-Moreno, I.; López, J.C. Access to 2,6-Dipropargylated BODIPYs as “Clickable” Congeners of Pyrromethene-567 Dye: Photostability and Synthetic Versatility. Org. Lett. 2021, 23, 6801–6806. [Google Scholar] [CrossRef]
- Blázquez-Moraleja, A.; Maierhofer, L.; Mann, E.; Prieto-Montero, R.; Oliden-Sánchez, A.; Celada, L.; Martínez-Martínez, V.; Chiara, M.-D.; Chiara, J.L. Acetoxymethyl-BODIPY dyes: A universal platform for the fluorescent labeling of nucleophiles. Org. Chem. Front. 2022, 9, 5774–5789. [Google Scholar] [CrossRef]
- Moerke, N.J. Fluorescence Polarization (FP) Assays for Monitoring Peptide-Protein or Nucleic Acid-Protein Binding. Curr. Protoc. Chem. Biol. 2009, 1, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. SLAS Discov. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Liu, H.; Shen, L.; Pan, C.; Huang, W. Structural modeling, energetic analysis and molecular design of a π-stacking system at the complex interface of pediatric respiratory syncytial virus nucleocapsid with the C-terminal peptide of phosphoprotein. Biophys. Chem. 2023, 292, 106916. [Google Scholar] [CrossRef]
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory Syncytial Virus: Virology, Reverse Genetics, and Pathogenesis of Disease. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–38. [Google Scholar] [CrossRef]
- Schobel, S.A.; Stucker, K.M.; Moore, M.L.; Anderson, L.J.; Larkin, E.K.; Shankar, J.; Bera, J.; Puri, V.; Shilts, M.H.; Rosas-Salazar, C.; et al. Respiratory Syncytial Virus whole-genome sequencing identifies convergent evolution of sequence duplication in the C-terminus of the G gene. Sci. Rep. 2016, 6, 26311. [Google Scholar] [CrossRef]
- Chapman, J.; Abbott, E.; Alber, D.G.; Baxter, R.C.; Bithell, S.K.; Henderson, E.A.; Carter, M.C.; Chambers, P.; Chubb, A.; Cockerill, G.S.; et al. RSV604, a novel inhibitor of respiratory syncytial virus replication. Antimicrob. Agents Chemother. 2007, 51, 3346–3353. [Google Scholar] [CrossRef]
- Challa, S.; Scott, A.D.; Yuzhakov, O.; Zhou, Y.; Tiong-Yip, C.L.; Gao, N.; Thresher, J.; Yu, Q. Mechanism of action for respiratory syncytial virus inhibitor RSV604. Antimicrob. Agents Chemother. 2015, 59, 1080–1087. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Rocchia, W.; Alexov, E.; Honig, B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions. J. Phys. Chem. B 2001, 105, 6507–6514. [Google Scholar] [CrossRef]
- Lu, D.; Teng, F.; Liu, Y.; Lu, L.; Chen, C.; Lei, J.; Wang, L.; Zhang, J. Self-assembly of magnetically recoverable ratiometric Cu2+ fluorescent sensor and adsorbent. RSC Adv. 2014, 4, 18660–18667. [Google Scholar] [CrossRef]
- Ng, C.Y.; Kwok, T.X.W.; Tan, F.C.K.; Low, C.-M.; Lam, Y. Fluorogenic probes to monitor cytosolic phospholipase A2 activity. Chem. Commun. 2017, 53, 1813–1816. [Google Scholar] [CrossRef]
- Gießler, K.; Griesser, H.; Göhringer, D.; Sabirov, T.; Richert, C. Synthesis of 3′-BODIPY-Labeled Active Esters of Nucleotides and a Chemical Primer Extension Assay on Beads. Eur. J. Org. Chem. 2010, 2010, 3611–3620. [Google Scholar] [CrossRef]
- Jullian, M.; Hernandez, A.; Maurras, A.; Puget, K.; Amblard, M.; Martinez, J.; Subra, G. N-terminus FITC labeling of peptides on solid support: The truth behind the spacer. Tetrahedron Lett. 2009, 50, 260–263. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Gonzalez Flecha, F.L.; Levi, V. Determination of the molecular size of BSA by fluorescence anisotropy. Biochem. Mol. Biol. Educ. 2003, 31, 319–322. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 708. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fn Peptide | Peptide Sequence | Kd (µM) | ΔFPmax (mFP) |
|---|---|---|---|
| F11 | ϕ-DS232DNDLS237LEDF-OH | 7.6 ± 0.9 | 130 ± 5 |
| F11EE | ϕ-DE232DNDLE237LEDF-OH | 0.38 ± 0.12/0.15 ± 0.12 (*) | 114 ± 6/93 ±13 (*) |
| F11SE | ϕ-DS232DNDLE237LEDF-OH | 0.87 ± 0.26/0.62 ± 0.25 (*) | 111 ± 9/97 ± 12 (*) |
| F9E | ϕ-DNDLE237LEDF-OH | 2.2 ± 0.3 | 148 ± 5 |
| F7E | ϕ-DLE237LEDF-OH | 2.5 ± 0.3 | 187 ± 7 |
| F5E | ϕ-E237LEDF-OH | 5.9 ± 0.5 | 285 ± 7 |
| F4 | ϕ-LEDF-OH | 35 ± 4 | 291 ± 20 |
| F3 | ϕ-EDF-OH | 62 ± 19 | 315 ± 61 |
| Fn Peptide | Inhibitor | IC50 (µM) | Ki (µM) | Kd of Fn Peptide (µM) * | Kd of Fn Equivalent to Inhibitor (µM) * |
|---|---|---|---|---|---|
| F11 | P11EE | 22 ± 4 | 6 | 7.6 | 0.38/0.15 |
| F11 | P9E | 66 ± 6 | 25 | 7.6 | 2.2 |
| F11 | P7E | 180 ± 11 | 75 | 7.6 | 2.5 |
| F11 | P11 | 86 ± 7 | 34 | 7.6 | 7.6 |
| F7E | P7E | 376 ± 26 | 74 | 2.5 | 2.5 |
| F5E | P5E | 433 ± 21 | 158 | 5.9 | 5.9 |
| F11 | M76 | 13 ± 2 | 3 | 7.6 | |
| F7E | M76 | 48 ± 5 | 8 | 2.5 |
| Fluorescent Molecule | Kd (µM) | M76 IC50 (µM) | M76 Ki (µM) |
|---|---|---|---|
| Fluorescein 1 | 91 ± 5 | 27 ± 2 | 13 |
| Fluorescein methyl ester 2 | 200 ± 8 | 46 ± 7 | 32 |
| Fluorescein ethyl ester 3 | 250 ± 27 | 65 ± 9 | 50 |
| 4,4′ difluoro BODIPY FL 4 | 870 ± 140 | (*) | (*) |
| Fluorescent BCNn Peptide | Fluorescent Peptide Sequence | ΔFPmax (mFP) | Kd (µM) | ΔFPmax (mFP) | Kd (µM) |
|---|---|---|---|---|---|
| No Salt | No Salt | With Salt | With Salt | ||
| BCN11SE | BCN-DSDNDLELEDF-OH | 312 ± 25 | 0.82 ± 0.22 | 122 ± 2 | 0.55 ± 0.07 |
| BCN11EE | BCN-DEDNDLELEDF-OH | 316 ± 12 | 0.28 ± 0.07 | 197 ± 5 | 0.26 ± 0.04 |
| BCN10EE | BCN-EDNDLELEDF-OH | 270 ± 7 | 0.16 ± 0.03 | 171 ± 16 | 1.24 ± 0.31 |
| BCN9E | BCN-DNDLELEDF-OH | 288 ± 15 | 0.96 ± 0.13 | ||
| BCN8E | BCN-NDLELEDF-OH | 298 ± 5 | 1.43 ± 0.08 | 284 ± 15 (*) | 7.1 ± 0.1 (*) |
| BCN7E | BCN-DLELEDF-OH | 332 ± 8 | 2.2 ± 0.2 | ||
| BCN6E | BCN-LELEDF-OH | 275 ± 8 | 2.3 ± 0.2 | 281 ± 19 (*) | 12.6 ± 1.6 (*) |
| BCN5E | BCN-ELEDF-OH | 431 ± 15 | 9.8 ± 0.9 |
| Fluorescent Peptide | Inhibitor | [N-RNA] (µM) | Fitted FPmax (mFP) | Fitted IC50 (µM) | Ki (µM) | Kd (µM) of BCN-P Peptide (*) |
|---|---|---|---|---|---|---|
| BCN10EE | P10EE | 1 | 214 | 21 ± 4 | 2.9 | 0.16 |
| BCN11EE | pP11 | 1 | 234 | 6.9 ± 1.2 | 1.4 | 0.28 |
| BCN11SE | M76 | 5 | 280 | 17 ± 2 | 1.8 | 0.8 |
| BCN10EE | M76 | 2 | 257 | 8.9 ± 0.6 | 0.5 | 0.16 |
| BCN10EE | M76 | 1 | 219 | 8.5 ± 0.6 | 1.1 | 0.16 |
| BCN8E | M76 | 5 | 196 | 13 ± 2 | 2.0 | 1.4 |
| BCN8E | M76 | 2 | 166 | 4.5 ± 0.4 | 1.3 | 1.4 |
| BCN6E | M76 | 10 | 177 | 14 ± 1 | 1.1 | 2.3 |
| BCN6E | M76 | 2 | 104 | 2.7 ± 0.2 | 0.7 | 2.3 |
| BCN5E | M76 | 20 | 280 | 26 ± 2 | 3.6 | 9.8 |
| BCN5E | M76 | 2 | 76 | 7.0 ± 0.6 | 4.8 | 9.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodjoyan, S.; Morissette, D.; Hontonnou, F.; Checa Ruano, L.; Richard, C.-A.; Sperandio, O.; Eléouët, J.-F.; Galloux, M.; Durand, P.; Deville-Foillard, S.; et al. Investigation of the Fuzzy Complex between RSV Nucleoprotein and Phosphoprotein to Optimize an Inhibition Assay by Fluorescence Polarization. Int. J. Mol. Sci. 2023, 24, 569. https://doi.org/10.3390/ijms24010569
Khodjoyan S, Morissette D, Hontonnou F, Checa Ruano L, Richard C-A, Sperandio O, Eléouët J-F, Galloux M, Durand P, Deville-Foillard S, et al. Investigation of the Fuzzy Complex between RSV Nucleoprotein and Phosphoprotein to Optimize an Inhibition Assay by Fluorescence Polarization. International Journal of Molecular Sciences. 2023; 24(1):569. https://doi.org/10.3390/ijms24010569
Chicago/Turabian StyleKhodjoyan, Silva, Deborha Morissette, Fortune Hontonnou, Luis Checa Ruano, Charles-Adrien Richard, Olivier Sperandio, Jean-François Eléouët, Marie Galloux, Philippe Durand, Stéphanie Deville-Foillard, and et al. 2023. "Investigation of the Fuzzy Complex between RSV Nucleoprotein and Phosphoprotein to Optimize an Inhibition Assay by Fluorescence Polarization" International Journal of Molecular Sciences 24, no. 1: 569. https://doi.org/10.3390/ijms24010569
APA StyleKhodjoyan, S., Morissette, D., Hontonnou, F., Checa Ruano, L., Richard, C.-A., Sperandio, O., Eléouët, J.-F., Galloux, M., Durand, P., Deville-Foillard, S., & Sizun, C. (2023). Investigation of the Fuzzy Complex between RSV Nucleoprotein and Phosphoprotein to Optimize an Inhibition Assay by Fluorescence Polarization. International Journal of Molecular Sciences, 24(1), 569. https://doi.org/10.3390/ijms24010569