
NRF2 in Cancer: Cross-Talk with Oncogenic Pathways and Involvement in Gammaherpesvirus-Driven Carcinogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NRF2 and Oncogenic Pathways
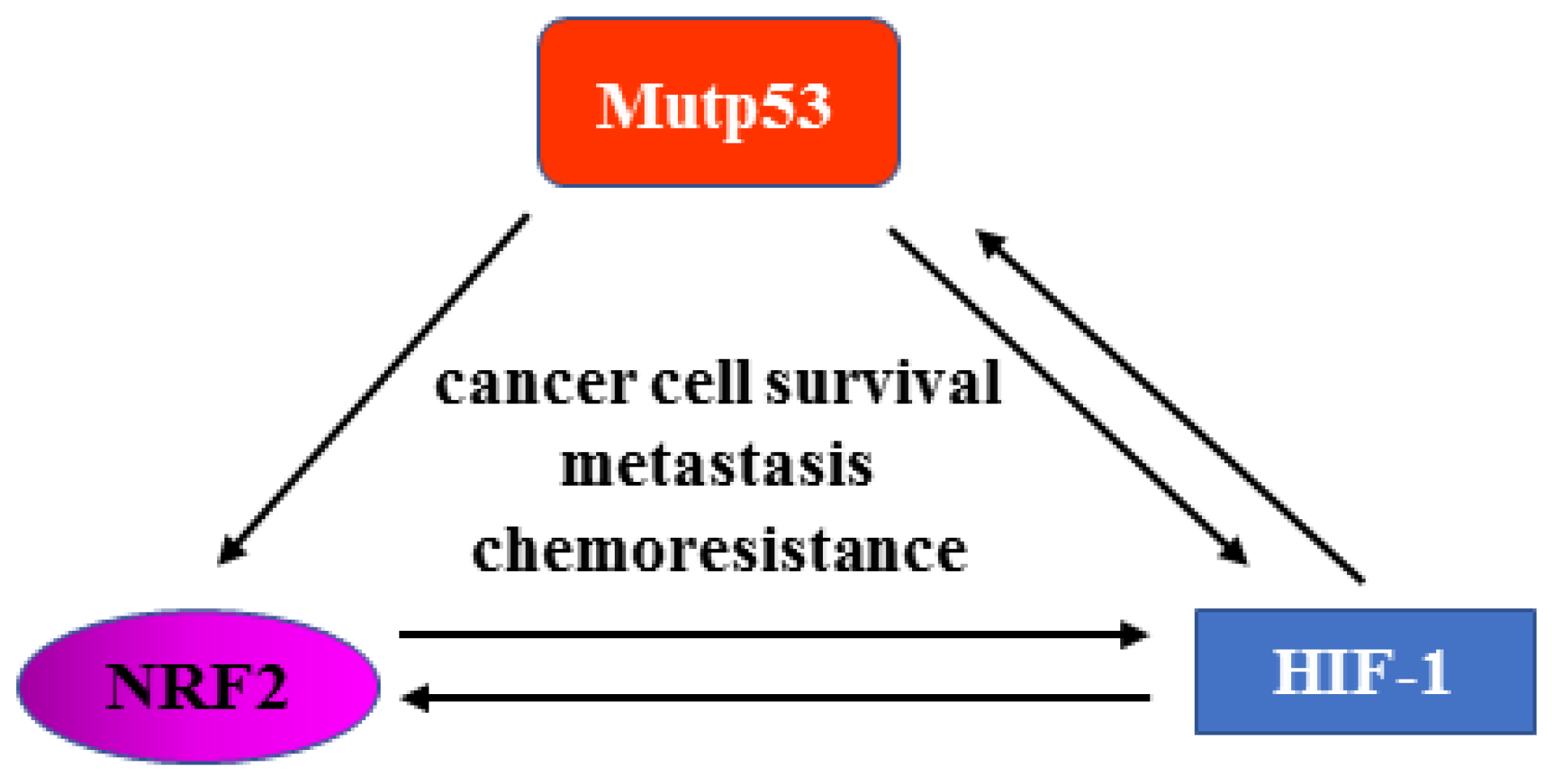
3. Interplay between NRF2 and p53
4. NRF2 and Gammaherpesvirus-Driven Cancers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, L.; Dong, H.; Song, E.; Xu, X.; Liu, L.; Song, Y. Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem. Biol. Interact. 2014, 209, 56–67. [Google Scholar] [CrossRef]
- Tripathi, D.; Nam, A.; Oldenburg, D.J.; Bendich, A.J. Reactive Oxygen Species, Antioxidant Agents, and DNA Damage in Developing Maize Mitochondria and Plastids. Front. Plant Sci. 2020, 11, 596. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmstrom, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinone, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox. Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Lv, Y.F.; Zhao, J.L.; You, Q.D.; Jiang, Z.Y. Regulation of Nrf2 by phosphorylation: Consequences for biological function and therapeutic implications. Free Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Dayalan Naidu, S.; Kostov, R.V.; Dinkova-Kostova, A.T. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol. Sci. 2015, 36, 6–14. [Google Scholar] [CrossRef]
- Home, T.; Jensen, R.A.; Rao, R. Heat shock factor 1 in protein homeostasis and oncogenic signal integration. Cancer Res. 2015, 75, 907–912. [Google Scholar] [CrossRef] [Green Version]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D’Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.M.; Hoskins, J.R.; Kravats, A.N.; Heffner, A.L.; Garikapati, S.; Wickner, S. Intermolecular Interactions between Hsp90 and Hsp70. J. Mol. Biol. 2019, 431, 2729–2746. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Huang, S.; Zhang, H.; Alikhan, M.; et al. Activation of MYC, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A.; Cirone, M. Mutant p53, Stabilized by Its Interplay with HSP90, Activates a Positive Feed-Back Loop Between NRF2 and p62 that Induces Chemo-Resistance to Apigenin in Pancreatic Cancer Cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Arena, A.; D’Orazi, G.; Cirone, M. VPA and TSA Interrupt the Interplay between mutp53 and HSP70, Leading to CHK1 and RAD51 Down-Regulation and Sensitizing Pancreatic Cancer Cells to AZD2461 PARP Inhibitor. Int. J. Mol. Sci. 2022, 23, 2268. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Baldari, S.; Pettinari, R.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Crispini, A.; Giorno, E.; Toietta, G.; Marchetti, F.; et al. A ruthenium(II)-curcumin compound modulates NRF2 expression balancing the cancer cell death/survival outcome according to p53 status. J. Exp. Clin. Cancer Res. 2020, 39, 122. [Google Scholar] [CrossRef]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Cirone, M. Cancer cells dysregulate PI3K/AKT/mTOR pathway activation to ensure their survival and proliferation: Mimicking them is a smart strategy of gammaherpesviruses. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 500–509. [Google Scholar] [CrossRef]
- Aramburu, J.; Ortells, M.C.; Tejedor, S.; Buxade, M.; Lopez-Rodriguez, C. Transcriptional regulation of the stress response by mTOR. Sci. Signal. 2014, 7, re2. [Google Scholar] [CrossRef]
- Shibata, T.; Saito, S.; Kokubu, A.; Suzuki, T.; Yamamoto, M.; Hirohashi, S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2-related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 2010, 70, 9095–9105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonnella, R.; Zarrella, R.; Santarelli, R.; Germano, C.A.; Gilardini Montani, M.S.; Cirone, M. Mechanisms of Sensitivity and Resistance of Primary Effusion Lymphoma to Dimethyl Fumarate (DMF). Int. J. Mol. Sci. 2022, 23, 6773. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Chiozzi, B.; Filardi, M.R.; Lotti, L.V.; Di Renzo, L.; Faggioni, A.; Cirone, M. Tyrosine kinase inhibitor tyrphostin AG490 triggers both apoptosis and autophagy by reducing HSF1 and Mcl-1 in PEL cells. Cancer Lett. 2015, 366, 191–197. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Traversi, G.; Gilardini Montani, M.S.; D’Orazi, V.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reduced chemotherapeutic sensitivity in high glucose condition: Implication of antioxidant response. Oncotarget 2019, 10, 4691–4702. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Giorno, E.; Gilardini Montani, M.S.; Pistritto, G.; Crispini, A.; Cirone, M.; D’Orazi, G. P62/SQSTM1/Keap1/NRF2 Axis Reduces Cancer Cells Death-Sensitivity in Response to Zn(II)-Curcumin Complex. Biomolecules 2021, 11, 348. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bail, Y. Nrf2 in cancer: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Kim, J.K.; Jang, H.D. Nrf2-mediated HO-1 induction coupled with the ERK signaling pathway contributes to indirect antioxidant capacity of caffeic acid phenethyl ester in HepG2 cells. Int. J. Mol. Sci. 2014, 15, 12149–12165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Liu, J.; Zhang, X.; Qi, J.; Yu, W.; Gu, Y. p38 MAPK-dependent Nrf2 induction enhances the resistance of glioma cells against TMZ. Med. Oncol. 2015, 32, 69. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A. Activation of Nrf2-antioxidant signaling attenuates NF-kB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Saeidi, S.; Cho, N.C.; Kim, S.H.; Lee, H.B.; Han, W.; Noh, D.Y.; Surh, Y.J. Interaction of Nrf2 with dimeric STAT3 induces IL-23 expression: Implications for breast cancer progression. Cancer Lett. 2021, 500, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cirone, M. Interconnected Adaptive Responses: A Way Out for Cancer Cells to Avoid Cellular Demise. Cancers 2022, 14, 2780. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY) 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotblat, B.; Melino, G.; Knight, R.A. NRF2 and p53: Januses in cancer? Oncotarget 2012, 3, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; D’Orazi, G. High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity. J. Exp. Clin. Cancer Res. 2014, 33, 79. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; Baldari, S.; Toietta, G.; Cirone, M.; D’Orazi, G. p53-Dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions. J. Exp. Clin. Cancer Res. 2017, 36, 126. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Garufi, A.; Cirone, M. Nuclear factor erythroid 2 (NF-E2) p45-related factor 2 interferes with homeodomain-interacting protein kinase 2/p53 activity to impair solid tumors chemosensitivity. IUBMB Life 2020, 72, 1634–1639. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef]
- Hofmann, T.G.; Glas, C.; Bitomsky, N. HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. Bioessays 2013, 35, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Torrente, L.; Sanchez, C.; Moreno, R.; Chowdhry, S.; Cabello, P.; Isono, K.; Koseki, H.; Honda, T.; Hayes, J.D.; Dinkova-Kostova, A.T.; et al. Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 2017, 36, 6204–6212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardinocchi, L.; Puca, R.; D’Orazi, G. HIF-1alpha antagonizes p53-mediated apoptosis by triggering HIPK2 degradation. Aging (Albany NY) 2011, 3, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhou, L.; Sun, X.; Li, Q. Homeodomain-interacting protein kinase 2 (HIPK2): A promising target for anti-cancer therapies. Oncotarget 2017, 8, 20452–20461. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Traversi, G.; Cirone, M.; D’Orazi, G. HIPK2 role in the tumor-host interaction: Impact on fibroblasts transdifferentiation CAF-like. IUBMB Life 2019, 71, 2055–2061. [Google Scholar] [CrossRef] [Green Version]
- Conte, A.; Valente, V.; Paladino, S.; Pierantoni, G.M. HIPK2 in cancer biology and therapy: Recent findings and future perspectives. Cell Signal. 2022, 101, 110491. [Google Scholar] [CrossRef]
- De la Vega, L.; Grishina, I.; Moreno, R.; Kruger, M.; Braun, T.; Schmitz, M.L. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol. Cell 2012, 46, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Wook Choi, D.; Yong Choi, C. HIPK2 modification code for cell death and survival. Mol. Cell Oncol. 2014, 1, e955999. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced NRF2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cirone, M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef] [Green Version]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [Green Version]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Taguchi, K.; Masamune, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Vakifahmetoglu-Norberg, H. Mutant p53 as a regulator and target of autophagy. Front. Oncol. 2021, 10, 607149. [Google Scholar] [CrossRef]
- Tang, Y.C.; Hsiao, J.R.; Jiang, S.S.; Chang, J.Y.; Chu, P.Y.; Liu, K.J.; Fang, H.L.; Lin, L.M.; Chen, H.H.; Huang, Y.W.; et al. c-MYC-directed NRF2 drives malignant progression of head and neck cancer via glucose-6-phosphate dehydrogenase and transketolase activation. Theranostics 2021, 11, 5232–5247. [Google Scholar] [CrossRef]
- Arena, A.; Gilardini Montani, M.S.; Romeo, M.A.; Benedetti, R.; Gaeta, A.; Cirone, M. DNA damage triggers an interplay between wtp53 and c-Myc affecting lymphoma cell proliferation and Kaposi sarcoma herpesvirus replication. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119168. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Gilardini Montani, M.S.; Arena, A.; Benedetti, R.; D’Orazi, G.; Cirone, M. c-Myc Sustains Pancreatic Cancer Cell Survival and mutp53 Stability through the Mevalonate Pathway. Biomedicines 2022, 10, 2489. [Google Scholar] [CrossRef]
- Gilardini Montani, M.S.; Tarquini, G.; Santarelli, R.; Gonnella, R.; Romeo, M.A.; Benedetti, R.; Arena, A.; Faggioni, A.; Cirone, M. p62/SQSTM1 promotes mitophagy and activates the NRF2-mediated antioxidant and anti-inflammatory response restraining EBV-driven B lymphocyte proliferation. Carcinogenesis 2022, 43, 277–287. [Google Scholar] [CrossRef]
- Gruosso, T.; Mieulet, V.; Cardon, M.; Bourachot, B.; Kieffer, Y.; Devun, F.; Dubois, T.; Dutreix, M.; Vincent-Salomon, A.; Miller, K.M.; et al. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Mol. Med. 2016, 8, 527–549. [Google Scholar] [CrossRef]
- Cirone, M. EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses 2018, 10, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjyshi, O.; Bottero, V.; Veettil, M.V.; Dutta, S.; Singh, V.V.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces Nrf2 during de novo infection of endothelial cells to create a microenvironment conducive to infection. PLoS Pathog. 2014, 10, e1004460. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Flaherty, S.; Veettil, M.V.; Johnson, K.E.; Chandran, B.; Bottero, V. Kaposi’s sarcoma-associated herpesvirus induces Nrf2 activation in latently infected endothelial cells through SQSTM1 phosphorylation and interaction with polyubiquitinated Keap1. J. Virol. 2015, 89, 2268–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemarie, Q.; Sugden, B. Epstein-Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef]
- Baquero-Perez, B.; Whitehouse, A. Hsp70 Isoforms Are Essential for the Formation of Kaposi’s Sarcoma-Associated Herpesvirus Replication and Transcription Compartments. PLoS Pathog. 2015, 11, e1005274. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; Gilardini Montani, M.S.; Angiolillo, C.; D’Orazi, G.; Faggioni, A.; Cirone, M. Cytotoxic Drugs Activate KSHV Lytic Cycle in Latently Infected PEL Cells by Inducing a Moderate ROS Increase Controlled by HSF1, NRF2 and p62/SQSTM1. Viruses 2018, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Gilardini Montani, M.S.; Santarelli, R.; Granato, M.; Gonnella, R.; Torrisi, M.R.; Faggioni, A.; Cirone, M. EBV reduces autophagy, intracellular ROS and mitochondria to impair monocyte survival and differentiation. Autophagy 2019, 15, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; Rizzello, C.; Romeo, M.A.; Yadav, S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Concomitant reduction of c-Myc expression and PI3K/AKT/mTOR signaling by quercetin induces a strong cytotoxic effect against Burkitt’s lymphoma. Int. J. Biochem. Cell Biol. 2016, 79, 393–400. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Gilardini Montani, M.S.; Cuomo, L.; Vitillo, M.; Santarelli, R.; Gonnella, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J. Nutr. Biochem. 2017, 41, 124–136. [Google Scholar] [CrossRef]
- Granato, M.; Gilardini Montani, M.S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef]
- Granato, M.; Gilardini Montani, M.S.; Filardi, M.; Faggioni, A.; Cirone, M. Capsaicin triggers immunogenic PEL cell death, stimulates DCs and reverts PEL-induced immune suppression. Oncotarget 2015, 6, 29543–29554. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirone, M.; D’Orazi, G. NRF2 in Cancer: Cross-Talk with Oncogenic Pathways and Involvement in Gammaherpesvirus-Driven Carcinogenesis. Int. J. Mol. Sci. 2023, 24, 595. https://doi.org/10.3390/ijms24010595
Cirone M, D’Orazi G. NRF2 in Cancer: Cross-Talk with Oncogenic Pathways and Involvement in Gammaherpesvirus-Driven Carcinogenesis. International Journal of Molecular Sciences. 2023; 24(1):595. https://doi.org/10.3390/ijms24010595
Chicago/Turabian StyleCirone, Mara, and Gabriella D’Orazi. 2023. "NRF2 in Cancer: Cross-Talk with Oncogenic Pathways and Involvement in Gammaherpesvirus-Driven Carcinogenesis" International Journal of Molecular Sciences 24, no. 1: 595. https://doi.org/10.3390/ijms24010595