
Modeling of Neurodegenerative Diseases: ‘Step by Step’ and ‘Network’ Organization of the Complexes of Model Systems
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Aging and the loss of ability to maintain normal processes of cell death and the utilization of abnormal proteins associated with aging. The most characteristic cases are taupathies (Alzheimer’s disease) and synucleinopathy (Parkinson’s disease);
- Mutations, most often expansion of trinucleotide repeats (Huntington’s disease; spinocerebellar ataxia types 1, 2, 3, 6, 7 and 17);
- Inflammatory processes in the nervous system;
- The effect of adverse environmental factors.
2. Basic Requirements for Models of Neurodegenerative Disorders
2.1. Variability and Phenotypic Plasticity
2.2. Biochemical Correspondence of the Model to the Organism under Study
2.3. The Rate of Reproduction and Model Response to Impact
2.4. Functional Correspondence of the Model to the Organism under Study
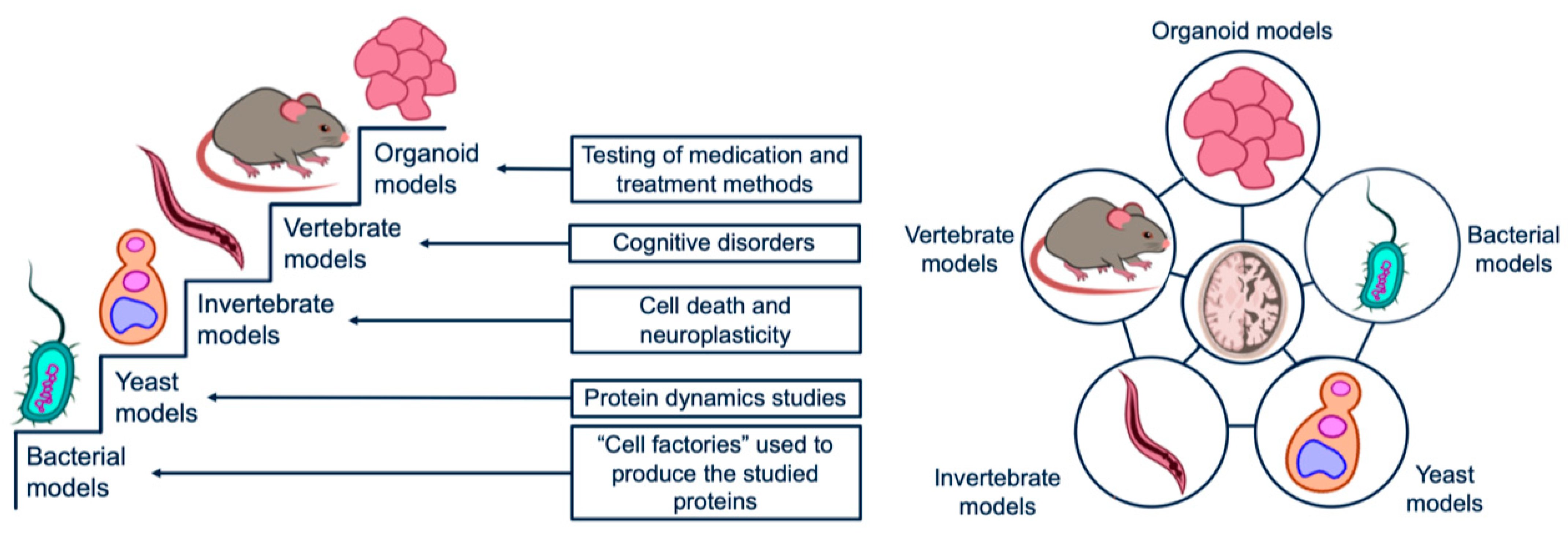
3. Prokaryotic Models
3.1. General Characteristics of Prokaryotic Models
3.2. The Role of the Intestine’s Microflora in the Development of Neurodegenerative Diseases: The ‘Microbiome-Brain-Gut’ Axis
- SHIME—The Simulator of the Human Intestinal Microbial Ecosystem. The system is a complex consisting of bioreactors connected in series with peristaltic pumps. In each bioreactor conditions specific to a particular section of the digestive system are created (an anaerobic environment and optimal temperature are maintained in all bioreactors). The reactors correspond to the following sections of the gastrointestinal tract: stomach, small intestine, ascending colon, transverse colon, and descending colon [27];
- SIMGI—SIMulator of GastroIntestinal tract. This system is similar to SHIME in many ways, but there are some differences. This model is not only fully automated but also controlled by a computer. Moreover, the gastric compartment has a peristaltic mixing system [26];
- Organ-On-A-Chip Systems. These include microfluidic systems, which are ultra-compact (within fractions of a millimeter) bioreactors containing organotypic cell cultures capable of reproducing the pathophysiological processes in miniature. In the case of modeling the ‘microbiome-brain-gut’ axis, the role of these systems is to reproduce the processes of the transfer of bacterial neurotoxins through various microanatomical barriers, including the blood-brain barrier [28];
3.3. Transgenic Bacterial Models
4. Yeast Models
4.1. The Role of Yeast Models in the Study of Neurodegenerative Diseases
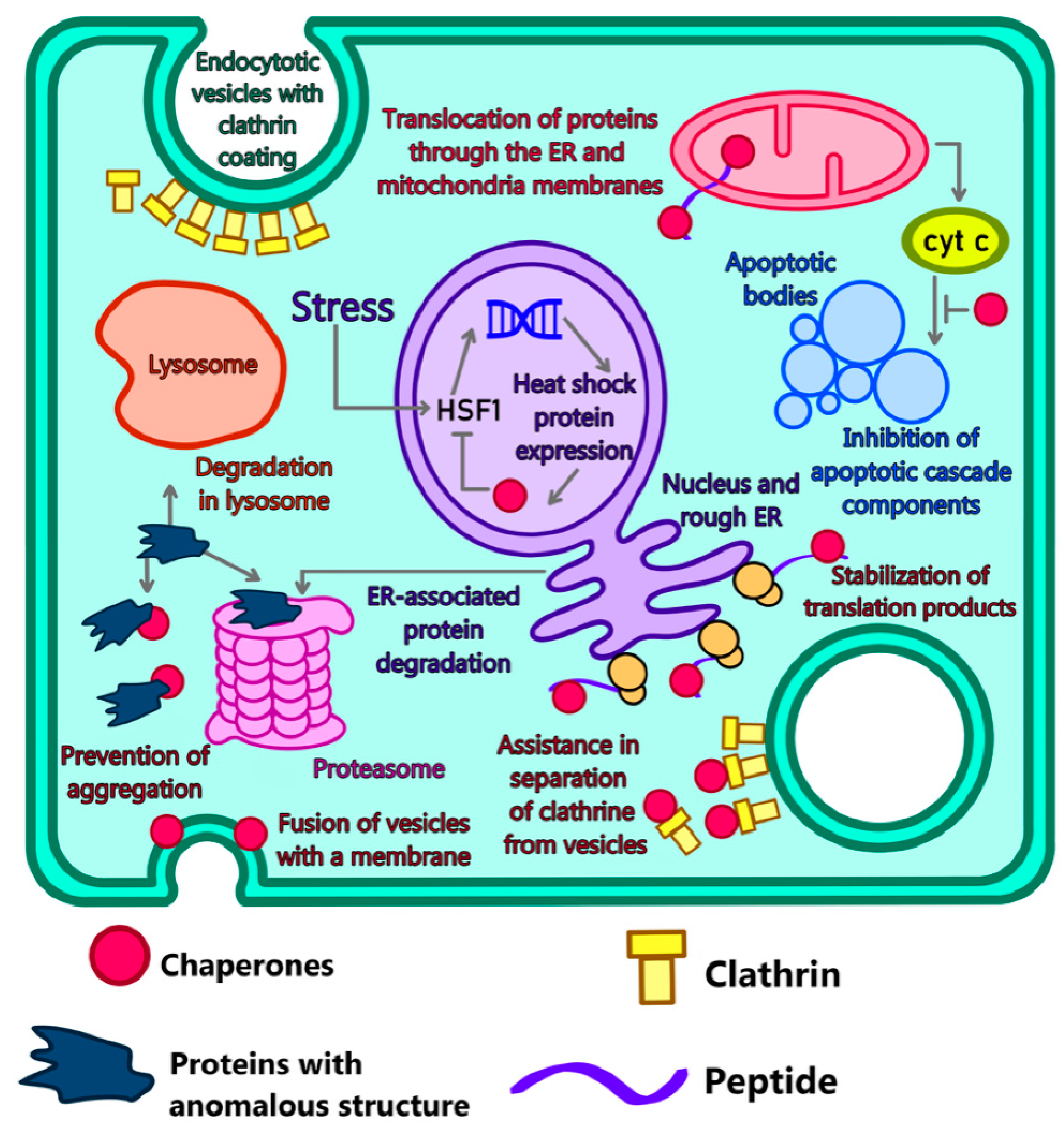
4.2. Modeling of Disorders in Chaperone Functioning
4.3. The role of Proteasomes and Their Interaction with Abnormal Proteins
4.4. Autophagy
5. Models Based on Invertebrates
5.1. Advantages of Multicellular Models
- The loss of the ability of cells and tissues to maintain normal cell death processes;
- Disorders in the utilization of misfolded and aggregated proteins that are toxic;
- The accumulation of cellular debris due to disorders of the autophagolysosomal apparatus;
- The uncontrolled course of inflammatory processes, mainly in the CNS.
- What does the toxic activity of abnormal protein aggregates look like at the molecular level?
- Is there a relationship between aggregation and toxicity?
- Do protein aggregates observed in neurodegenerative diseases have any common properties, including pathological ones?
- Why are these diseases associated with aging?
- What causes the development of pathologies associated with a particular cell type [50]?
5.2. Invertebrates as Models for Neurodegenerative Disorders
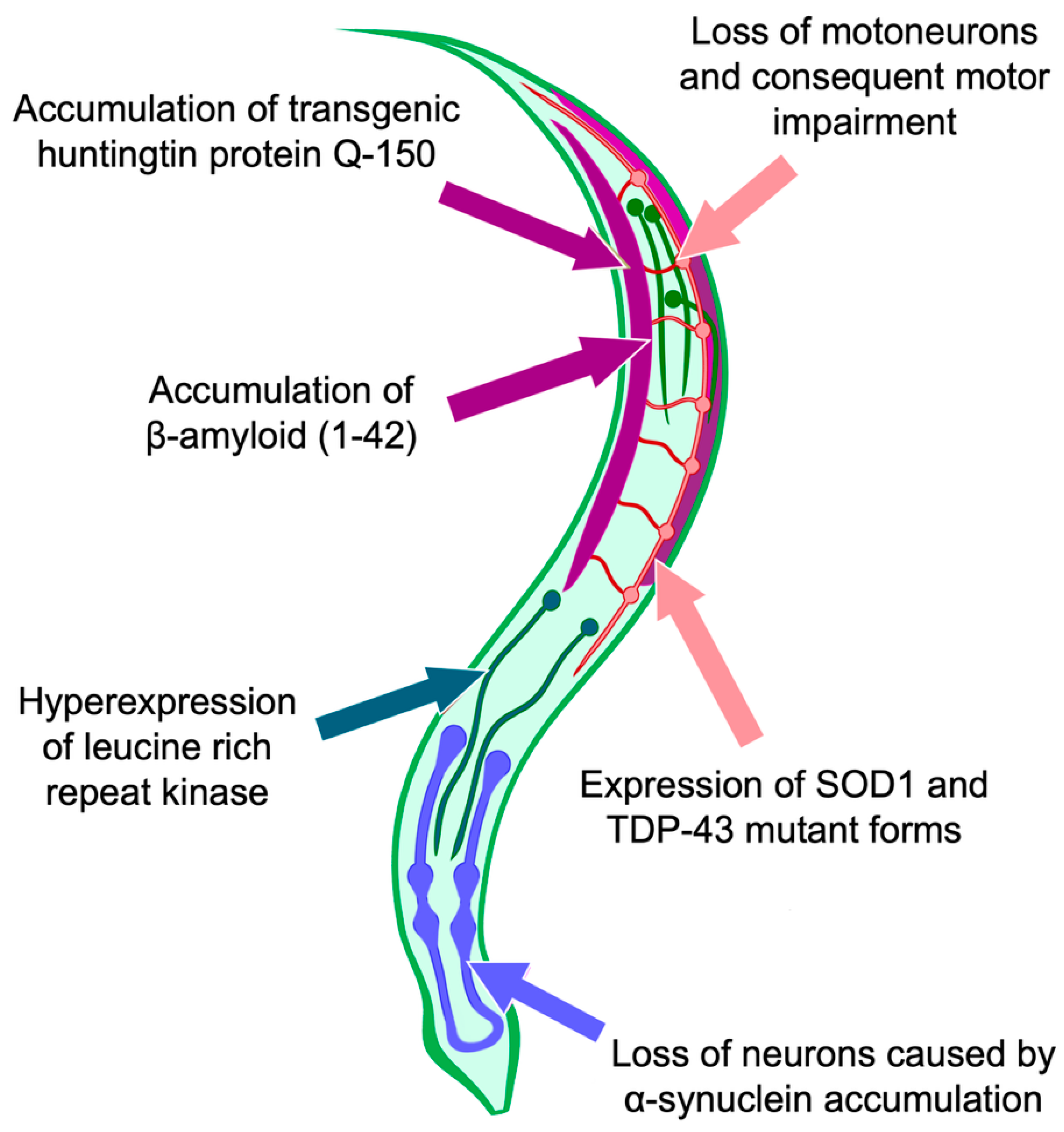
5.3. Use of C. elegans as a Model for Studying the Processes of Cell Death and Autophagy
6. Mammals-Based Models
6.1. Advantages of Using Mammals
- The necessity to study the molecular mechanisms and mechanisms of initiation of proteinopathy. The neurodegenerative process can be considered in its entirety, considering the influence of all organs and systems of the body, even if phenotypically the degeneration is not fully manifested;
- The relative ease of maintaining the model system;
- The presence of age-related changes as a natural process inherent in the body of an animal. Aging, being a systemic process of reducing the body’s ability to maintain its functional state, is the main risk factor for a number of diseases, including neurodegenerative ones;
- Highly organized immune system;
- Presence of complex neural activity and behavior (especially in primate-based models).
6.2. The Use of Mammals to Model Aging Processes
6.3. The study of Oxidative Stress Processes as a Factor in the Development of Neurodegenerative Diseases
6.4. The Use of Mammals to Study the Role of the Immune System in the Neuropathology Development
6.5. Behavioral Disorders Modeling
7. Organoid Models of Neurodegenerative Diseases
7.1. General Characteristics of Organoid Models
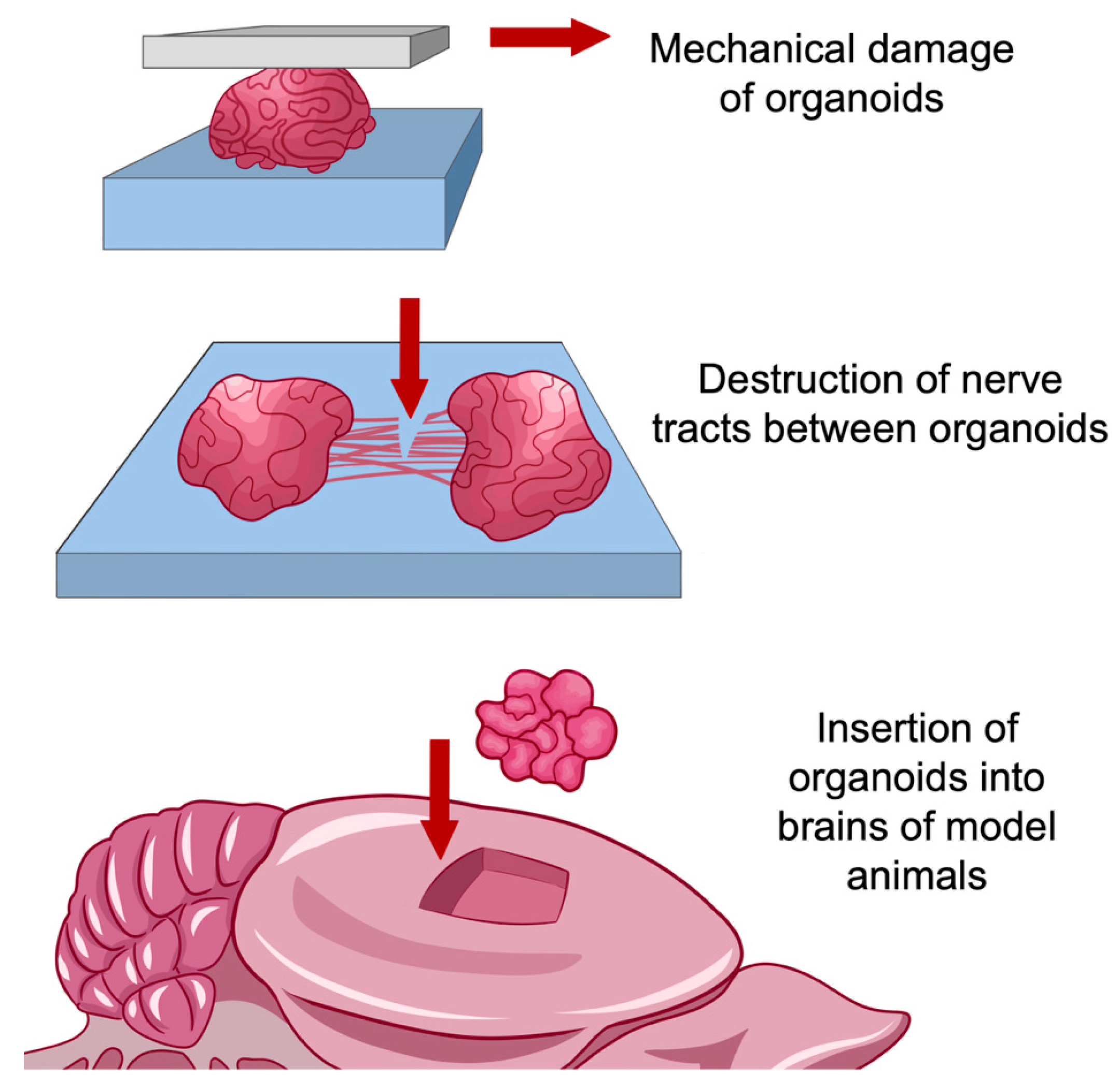
7.2. Use of Organoid Models for Research in the Field of Regeneration and Traumatology
- The impossibility of reliable reproduction of inflammatory and degenerative processes associated with brain injuries in animal models, especially in rodents. In addition to differences between humans and model animals at the molecular level, interspecies differences in brain structure are a significant factor, such as different ratios of gray and white matter, the density of neurons in the cortex, and a number of other structural differences [80]. Simulation accuracy is improved when using large animals such as pigs or primates. However, in this case, the duration of the experiment and its resource consumption increase significantly;
- The ability to fully trace the cell path, from pluripotent stem cells to nerve and glial cells, so that changes in the architecture of the nervous tissue could be observed in real-time. In most methods of working with animal models, only a certain time ‘slice’ of structures is considered, corresponding to their state at a certain point in time;
- Creation within the model of niches with a unique microenvironment. The cell cultures used to model the long-term effects of brain injury prior to the introduction of organoid models into practice had a quite simple structure. For the most part, these are single-layer or multilayer structures containing one or more cell types [81]. Organoid models, due to their complex structure, are able to reproduce the microenvironment specific to cells in the composition of an organ, so they have greater potential for research compared to cell cultures [82,83].
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wojaczyńska-Stanek, K.; Adamek, D.; Marszał, E.; Hoffman-Zacharska, D. Huntington disease in a 9-year-old boy: Clinical course and neuropathologic examination. J. Child Neurol. 2006, 21, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet. Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Liu, Y.; Gerasimov, E.S.; Vattathil, S.M.; Wynne, M.E.; Liu, J.; Lori, A.; Faundez, V.; Bennett, D.A.; Seyfried, N.T.; et al. Shared mechanisms across the major psychiatric and neurodegenerative diseases. Nat. Commun. 2022, 13, 4314. [Google Scholar] [CrossRef]
- MacMillan, J.C.; Snell, R.G.; Tyler, A.; Houlihan, G.D.; Fenton, I.; Cheadle, J.P.; Lazarou, L.P.; Shaw, D.J.; Harper, P.S. Molecular analysis and clinical correlations of the Huntington’s disease mutation. Lancet (Lond. Engl.) 1993, 342, 954–958. [Google Scholar] [CrossRef]
- Grenier, K.; Kao, J.; Diamandis, P. Three-dimensional modeling of human neurodegeneration: Brain organoids coming of age. Mol. Psychiatry 2020, 25, 254–274. [Google Scholar] [CrossRef] [PubMed]
- Cascione, M.; De Matteis, V.; Leporatti, S.; Rinaldi, R. The New Frontiers in Neurodegenerative Diseases Treatment: Liposomal-Based Strategies. Front. Bioeng. Biotechnol. 2020, 8, 566767. [Google Scholar] [CrossRef] [PubMed]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Cai, L.; Huang, J. Schizophrenia and risk of dementia: A meta-analysis study. Neuropsychiatr. Dis. Treat. 2018, 14, 2047–2055. [Google Scholar] [CrossRef] [Green Version]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Manciocco, A.; Chiarotti, F.; Vitale, A.; Calamandrei, G.; Laviola, G.; Alleva, E. The application of Russell and Burch 3R principle in rodent models of neurodegenerative disease: The case of Parkinson’s disease. Neurosci. Biobehav. Rev. 2009, 33, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Savonenko, A.; Schilling, G.; Wang, J.; Xu, G.; Borchelt, D.R. Transgenic mouse models of neurodegenerative disease: Opportunities for therapeutic development. Curr. Neurol. Neurosci. Rep. 2002, 2, 457–464. [Google Scholar] [CrossRef]
- Huillard d’Aignaux, J.; Costagliola, D.; Maccario, J.; Billette de Villemeur, T.; Brandel, J.P.; Deslys, J.P.; Hauw, J.J.; Chaussain, J.L.; Agid, Y.; Dormont, D.; et al. Incubation period of Creutzfeldt-Jakob disease in human growth hormone recipients in France. Neurology 1999, 53, 1197–1201. [Google Scholar] [CrossRef]
- Mackenzie, G.; Will, R. Creutzfeldt-Jakob disease: Recent developments. F1000Research 2017, 6, 2053. [Google Scholar] [CrossRef]
- Puderbaugh, M.; Emmady, P.D. Neuroplasticity; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Raimondi, I.; Izzo, L.; Tunesi, M.; Comar, M.; Albani, D.; Giordano, C. Organ-On-A-Chip in vitro Models of the Brain and the Blood-Brain Barrier and Their Value to Study the Microbiota-Gut-Brain Axis in Neurodegeneration. Front. Bioeng. Biotechnol. 2019, 7, 435. [Google Scholar] [CrossRef]
- Friedland, R.P.; McMillan, J.D.; Kurlawala, Z. What Are the Molecular Mechanisms by Which Functional Bacterial Amyloids Influence Amyloid Beta Deposition and Neuroinflammation in Neurodegenerative Disorders? Int. J. Mol. Sci. 2020, 21, 1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulusoy, A.; Phillips, R.J.; Helwig, M.; Klinkenberg, M.; Powley, T.L.; Di Monte, D.A. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol. 2017, 133, 381–393. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Ceppa, F.A.; Izzo, L.; Sardelli, L.; Raimondi, I.; Tunesi, M.; Albani, D.; Giordano, C. Human Gut-Microbiota Interaction in Neurodegenerative Disorders and Current Engineered Tools for Its Modeling. Front. Cell. Infect. Microbiol. 2020, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Van de Wiele, T.; Van den Abbeele, P.; Ossieur, W.; Possemiers, S.; Marzorati, M. The Simulator of the Human Intestinal Microbial Ecosystem (SHIME®). In The Impact of Food Bioactives on Health; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Swtzerland, 2015; pp. 305–317. ISBN 978-3-319-15791-7. [Google Scholar]
- Raimondi, M.T.; Albani, D.; Giordano, C. An Organ-On-A-Chip Engineered Platform to Study the Microbiota-Gut-Brain Axis in Neurodegeneration. Trends Mol. Med. 2019, 25, 737–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Miralles, N.; Villaverde, A. Bacterial cell factories for recombinant protein production; expanding the catalogue. Microb. Cell Fact. 2013, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Legname, G.; Baskakov, I.V.; Nguyen, H.-O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef]
- Sherman, M.Y.; Muchowski, P.J. Making yeast tremble: Yeast models as tools to study neurodegenerative disorders. Neuromol. Med. 2003, 4, 133–146. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.I.; Muchowski, P.J. Small heat-shock proteins and their potential role in human disease. Curr. Opin. Struct. Biol. 2000, 10, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, K.; Baker, H.F.; Crow, T.J.; Poulter, M.; Owen, F.; Terwilliger, J.D.; Westaway, D.; Ott, J.; Prusiner, S.B. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature 1989, 338, 342–345. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Masison, D.C. Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept. Lett. 2009, 16, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- García-Martínez, M.Á.; Montejo González, J.C.; García-de-Lorenzo y Mateos, A.; Teijeira, S. Muscle weakness: Understanding the principles of myopathy and neuropathy in the critically ill patient and the management options. Clin. Nutr. 2020, 39, 1331–1344. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cebollero, E.; Reggiori, F. Regulation of autophagy in yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2009, 1793, 1413–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Hale, A.N.; Ledbetter, D.J.; Gawriluk, T.R.; Rucker, E.B. 3rd Autophagy: Regulation and role in development. Autophagy 2013, 9, 951–972. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trancikova, A.; Tsika, E.; Moore, D.J. Mitochondrial dysfunction in genetic animal models of Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 896–919. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.J.; Scheibye-Knudsen, M.; Longo, D.L.; de Cabo, R. Animal models of aging research: Implications for human aging and age-related diseases. Annu. Rev. Anim. Biosci. 2015, 3, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Link, C.D. Invertebrate models of Alzheimer’s disease. Genes. Brain. Behav. 2005, 4, 147–156. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Glial cell reactions in neurodegenerative diseases: Pathophysiology and therapeutic interventions. Alzheimer Dis. Assoc. Disord. 1998, 12 (Suppl. 2), S1–S6. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Strobusch, A.D.; Jefferson, J.W. The checkered history of lithium in medicine. Pharm. Hist. 1980, 22, 72–76. [Google Scholar] [PubMed]
- Phiel, C.J.; Klein, P.S. Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 789–813. [Google Scholar] [CrossRef] [PubMed]
- Jans, K.; Lüersen, K.; Rimbach, G. Drosophila melanogaster as a Model Organism to Study Lithium and Boron Bioactivity. Int. J. Mol. Sci. 2021, 22, 1710. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, T.C. Signal transduction by the Wnt family of ligands. Biochem. J. 1998, 329 Pt 2, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Shruster, A.; Eldar-Finkelman, H.; Melamed, E.; Offen, D. Wnt signaling pathway overcomes the disruption of neuronal differentiation of neural progenitor cells induced by oligomeric amyloid β-peptide. J. Neurochem. 2011, 116, 522–529. [Google Scholar] [CrossRef]
- Yao, C.; El Khoury, R.; Wang, W.; Byrd, T.A.; Pehek, E.A.; Thacker, C.; Zhu, X.; Smith, M.A.; Wilson-Delfosse, A.L.; Chen, S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Tilleman, L.; Germani, F.; De Henau, S.; Geuens, E.; Hoogewijs, D.; Braeckman, B.P.; Vanfleteren, J.R.; Moens, L.; Dewilde, S. Globins in Caenorhabditis elegans. IUBMB Life 2011, 63, 166–174. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef]
- Cornaglia, M.; Krishnamani, G.; Mouchiroud, L.; Sorrentino, V.; Lehnert, T.; Auwerx, J.; Gijs, M.A.M. Automated longitudinal monitoring of in vivo protein aggregation in neurodegenerative disease C. elegans models. Mol. Neurodegener. 2016, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, P.E.A.; Zhang, Y.-J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef] [PubMed]
- Baskoylu, S.N.; Yersak, J.; O’Hern, P.; Grosser, S.; Simon, J.; Kim, S.; Schuch, K.; Dimitriadi, M.; Yanagi, K.S.; Lins, J.; et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018, 14, e1007682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. (Elite Ed.) 2013, 5, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Spangler, E.L.; Ingram, D.K. Utilization of the rat as a model of mammalian aging: Impact of pathology on behavior. Gerontology 1996, 42, 301–311. [Google Scholar] [CrossRef]
- Flurkey, K.; Currer, J.M.; Harrison, D.E. Mouse Models in Aging Research. In The Mouse in Biomedical Research; Fox, J.G., Davisson, M.T., Quimby, F.W., Barthold, S.W., Newcomer, C.E., Smith, A.L., Eds.; American College of Laboratory Animal Medicine; Elsevier: Burlington, VT, USA, 2007; pp. 637–672. ISBN 978-0-12-369454-6. [Google Scholar]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Pilotto, A.; Padovani, A.; Borroni, B. Clinical, biological, and imaging features of monogenic Alzheimer’s Disease. Biomed Res. Int. 2013, 2013, 689591. [Google Scholar] [CrossRef] [Green Version]
- Pihlstrøm, L.; Wiethoff, S.; Houlden, H. Genetics of neurodegenerative diseases: An overview. Handb. Clin. Neurol. 2017, 145, 309–323. [Google Scholar] [CrossRef]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar] [CrossRef]
- Svensson, M.; Lexell, J.; Deierborg, T. Effects of Physical Exercise on Neuroinflammation, Neuroplasticity, Neurodegeneration, and Behavior: What We Can Learn From Animal Models in Clinical Settings. Neurorehabil. Neural Repair 2015, 29, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sephton, C.F.; Good, S.K.; Atkin, S.; Dewey, C.M.; Mayer, P., 3rd; Herz, J.; Yu, G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 2010, 285, 6826–6834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.-S.; Cheng, W.-C.; Hou, S.-C.; Yan, Y.-T.; Jiang, S.-T.; Shen, C.-K.J. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis 2010, 48, 56–62. [Google Scholar] [CrossRef]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef]
- Khaitovich, P.; Muetzel, B.; She, X.; Lachmann, M.; Hellmann, I.; Dietzsch, J.; Steigele, S.; Do, H.-H.; Weiss, G.; Enard, W.; et al. Regional patterns of gene expression in human and chimpanzee brains. Genome Res. 2004, 14, 1462–1473. [Google Scholar] [CrossRef] [Green Version]
- Semendeferi, K.; Teffer, K.; Buxhoeveden, D.P.; Park, M.S.; Bludau, S.; Amunts, K.; Travis, K.; Buckwalter, J. Spatial organization of neurons in the frontal pole sets humans apart from great apes. Cereb. Cortex 2011, 21, 1485–1497. [Google Scholar] [CrossRef] [Green Version]
- Jgamadze, D.; Johnson, V.E.; Wolf, J.A.; Cullen, D.K.; Song, H.; Ming, G.-L.; Smith, D.H.; Chen, H.I. Modeling traumatic brain injury with human brain organoids. Curr. Opin. Biomed. Eng. 2020, 14, 52–58. [Google Scholar] [CrossRef]
- Pasqualini, C.; Kozaki, T.; Bruschi, M.; Nguyen, T.H.H.; Minard-Colin, V.; Castel, D.; Grill, J.; Ginhoux, F. Modeling the Interaction between the Microenvironment and Tumor Cells in Brain Tumors. Neuron 2020, 108, 1025–1044. [Google Scholar] [CrossRef]
- Xia, T.; Du, W.-L.; Chen, X.-Y.; Zhang, Y.-N. Organoid models of the tumor microenvironment and their applications. J. Cell. Mol. Med. 2021, 25, 5829–5841. [Google Scholar] [CrossRef]
- Azzarelli, R. Organoid Models of Glioblastoma to Study Brain Tumor Stem Cells. Front. cell Dev. Biol. 2020, 8, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From inflammation to cancer. Immun. Ageing 2018, 15, 1. [Google Scholar] [CrossRef]
- Rong, L.L.; Gooch, C.; Szabolcs, M.; Herold, K.C.; Lalla, E.; Hays, A.P.; Yan, S.F.; Yan, S.S.D.; Schmidt, A.M. RAGE: A journey from the complications of diabetes to disorders of the nervous system—Striking a fine balance between injury and repair. Restor. Neurol. Neurosci. 2005, 23, 355–365. [Google Scholar] [PubMed]
- Jiang, X.; Wang, X.; Tuo, M.; Ma, J.; Xie, A. RAGE and its emerging role in the pathogenesis of Parkinson’s disease. Neurosci. Lett. 2018, 672, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thornalley, P.J. Advanced glycation endproducts: What is their relevance to diabetic complications? Diabetes. Obes. Metab. 2007, 9, 233–245. [Google Scholar] [CrossRef]
- Grillo, M.A.; Colombatto, S. Advanced glycation end-products (AGEs): Involvement in aging and in neurodegenerative diseases. Amino Acids 2008, 35, 29–36. [Google Scholar] [CrossRef]
- Ambrosini, Y.M.; Borcherding, D.; Kanthasamy, A.; Kim, H.J.; Willette, A.A.; Jergens, A.; Allenspach, K.; Mochel, J.P. The Gut-Brain Axis in Neurodegenerative Diseases and Relevance of the Canine Model: A Review. Front. Aging Neurosci. 2019, 11, 130. [Google Scholar] [CrossRef]
- Kingsbury, D.D.; Sun, L.; Qi, Y.; Fredericks, J.; Wang, Q.; Wannemuehler, M.J.; Jergens, A.; Allenspach, K. Optimizing the Development and Characterization of Canine Small Intestinal Crypt Organoids as a Research Model. Gastroenterology 2017, 152, S353. [Google Scholar] [CrossRef]
- Mochel, J.P.; Jergens, A.E.; Kingsbury, D.; Kim, H.J.; Martín, M.G.; Allenspach, K. Intestinal Stem Cells to Advance Drug Development, Precision, and Regenerative Medicine: A Paradigm Shift in Translational Research. AAPS J. 2017, 20, 17. [Google Scholar] [CrossRef] [Green Version]
- Chandra, L.; Borcherding, D.C.; Kingsbury, D.; Atherly, T.; Ambrosini, Y.M.; Bourgois-Mochel, A.; Yuan, W.; Kimber, M.; Qi, Y.; Wang, Q.; et al. Derivation of adult canine intestinal organoids for translational research in gastroenterology. BMC Biol. 2019, 17, 33. [Google Scholar] [CrossRef]
- Shcheglovitov, A.; Peterson, R.T. Screening Platforms for Genetic Epilepsies-Zebrafish, iPSC-Derived Neurons, and Organoids. Neurother. J. Am. Soc. Exp. Neurother. 2021, 18, 1478–1489. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasko, V.I.; Churkina, A.S.; Shakhov, A.S.; Kotlobay, A.A.; Alieva, I.B. Modeling of Neurodegenerative Diseases: ‘Step by Step’ and ‘Network’ Organization of the Complexes of Model Systems. Int. J. Mol. Sci. 2023, 24, 604. https://doi.org/10.3390/ijms24010604
Pasko VI, Churkina AS, Shakhov AS, Kotlobay AA, Alieva IB. Modeling of Neurodegenerative Diseases: ‘Step by Step’ and ‘Network’ Organization of the Complexes of Model Systems. International Journal of Molecular Sciences. 2023; 24(1):604. https://doi.org/10.3390/ijms24010604
Chicago/Turabian StylePasko, Viacheslav Igorevich, Aleksandra Sergeevna Churkina, Anton Sergeevich Shakhov, Anatoly Alexeevich Kotlobay, and Irina Borisovna Alieva. 2023. "Modeling of Neurodegenerative Diseases: ‘Step by Step’ and ‘Network’ Organization of the Complexes of Model Systems" International Journal of Molecular Sciences 24, no. 1: 604. https://doi.org/10.3390/ijms24010604
APA StylePasko, V. I., Churkina, A. S., Shakhov, A. S., Kotlobay, A. A., & Alieva, I. B. (2023). Modeling of Neurodegenerative Diseases: ‘Step by Step’ and ‘Network’ Organization of the Complexes of Model Systems. International Journal of Molecular Sciences, 24(1), 604. https://doi.org/10.3390/ijms24010604