
Molecular Mechanisms Underlying IL-33-Mediated Inflammation in Inflammatory Bowel Disease




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
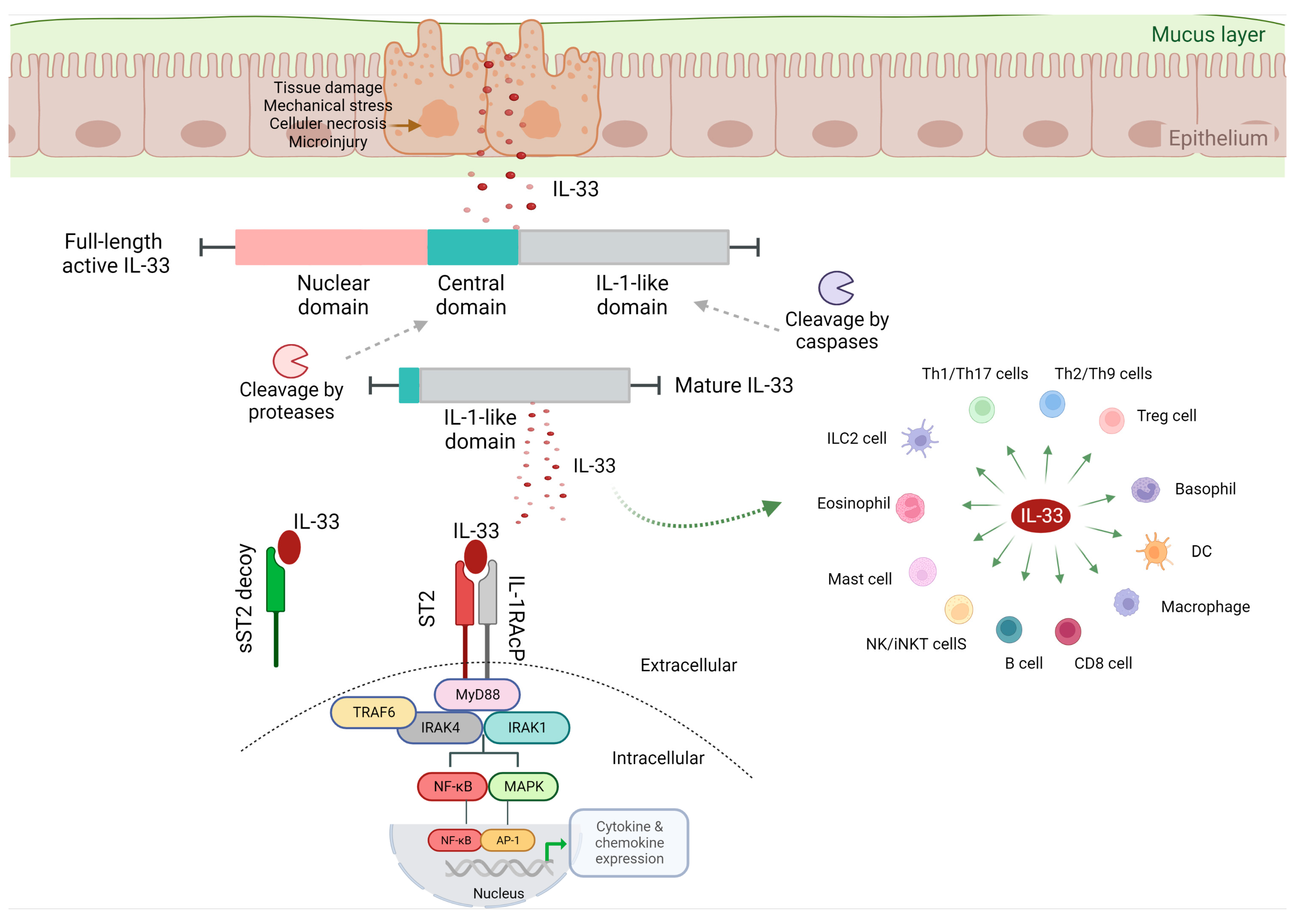
2. IL-33 Biological Function
2.1. IL-33/ST2 Signaling
2.2. IL-33/ST2 Signaling and Mucosal Immunity
3. IL-33 and IBD
4. IL-33 and Ulcerative Colitis
4.1. IL-33 Expression in UC-Affected Tissues
4.2. Detrimental Role of IL-33
4.3. Protective Role of IL-33
5. IL-33 and Crohn’s Disease
6. IL-33 and Intestinal Fibrosis
7. Conclusions
8. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [PubMed]
- Kofla-Dłubacz, A.; Pytrus, T.; Akutko, K. Etiology of IBD-Is It Still a Mystery? Int. J. Mol. Sci. 2022, 23, 12445. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef]
- Cobrin, G.M.; Abreu, M.T. Defects in mucosal immunity leading to Crohn’s disease. Immunol. Rev. 2005, 206, 277–295. [Google Scholar] [CrossRef]
- Targan, S.R.; Karp, L.C. Defects in mucosal immunity leading to ulcerative colitis. Immunol. Rev. 2005, 206, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Schirbel, A.; Fiocchi, C. Inflammatory bowel disease: Established and evolving considerations on its etiopathogenesis and therapy. J. Dig. Dis. 2010, 11, 266–276. [Google Scholar] [PubMed]
- Burrelli Scotti, G.; Afferri, M.T.; De Carolis, A.; Vaiarello, V.; Fassino, V.; Ferrone, F.; Minisola, S.; Nieddu, L.; Vernia, P. Factors affecting vitamin D deficiency in active inflammatory bowel diseases. Dig. Liver. Dis. 2019, 51, 657–662. [Google Scholar] [CrossRef]
- Rosen, C.E.; Palm, N.W. Navigating the microbiota seas: Triangulation finds a way forward. Cell Host Microbe 2018, 23, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Gkouskou, K.K.; Deligianni, C.; Tsatsanis, C.; Eliopoulos, A.G. The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell Infect Microbiol. 2014, 4, 28. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.A.; O’Callaghan, A.; Corr, S.C. IL-33 and IL-18 in Inflammatory Bowel Disease Etiology and Microbial Interactions. Front. Immunol. 2019, 10, 1091. [Google Scholar] [CrossRef] [Green Version]
- Andoh, A.; Nishida, A. Pro- and anti-inflammatory roles of interleukin (IL)-33, IL-36, and IL-38 in inflammatory bowel disease. J. Gastroenterol. 2022; ahead of print. [Google Scholar] [CrossRef]
- Beltrán, C.J.; Núñez, L.E.; Díaz-Jiménez, D.; Farfan, N.; Candia, E.; Heine, C.; López, F.; González, M.J.; Quera, R.; Hermoso, M.A. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm. Bowel. Dis. 2010, 16, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Jiménez, D.; De la Fuente, M.; Dubois-Camacho, K.; Landskron, G.; Fuentes, J.; Pérez, T.; González, M.J.; Simian, D.; Hermoso, M.A.; Quera, R. Soluble ST2 is a sensitive clinical marker of ulcerative colitis evolution. BMC Gastroenterol. 2016, 16, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorelli, L.; Garg, R.R.; Hoang, S.B.; Spina, L.; Mattioli, B.; Scarpa, M.; Fiocchi, C.; Vecchi, M.; Pizarro, T.T. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci USA 2010, 107, 8017–8022. [Google Scholar] [CrossRef] [Green Version]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef]
- Murdaca, G.; Greco, M.; Tonacci, A. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayter, S.M.; Cook, M.C. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun. Rev. 2012, 11, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Hodzic, Z.; Schill, E.M.; Bolock, A.M.; Good, M. IL-33 and the intestine: The good, the bad, and the inflammatory. Cytokine 2017, 100, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves-Filho, J.C.; Sônego, F.; Souto, F.O.; Freitas, A.; Verri, W.A., Jr.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Smithgall, M.D.; Comeau, M.R.; Yoon, B.R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.P. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef]
- Guo, H.; Bossila, E.A. Dual Immune Regulatory Roles of Interleukin-33 in Pathological Conditions. Cells 2022, 11, 3237. [Google Scholar] [CrossRef]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Pastorelli, L.; De Salvo, C.; Cominelli, M.A.; Vecchi, M.; Pizarro, T.T. Novel cytokine signaling pathways in inflammatory bowel disease: Insight into the dichotomous functions of IL-33 during chronic intestinal inflammation. Ther. Adv. Gastroenterol. 2011, 4, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrançais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [Green Version]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Rajput, C.; Hong, J.Y.; Lei, J.; Hinde, J.L. The Innate Cytokines IL-25, IL-33, and TSLP Cooperate in the Induction of Type 2 Innate Lymphoid Cell Expansion and Mucous Metaplasia in Rhinovirus-Infected Immature Mice. J. Immunol. 2017, 199, 1308–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Yin, H.; Yuan, B.; Liu, T.; Luo, L.; Huang, P.; Dai, L.; Zeng, K. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Mol. Immunol. 2017, 90, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Moussion, C.; Ortega, N.; Girard, J.P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel ‘alarmin’? PLoS One 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Oshio, T.; Komine, M.; Tsuda, H.; Tominaga, S.I.; Saito, H.; Nakae, S.; Ohtsuki, M. Nuclear expression of IL-33 in epidermal keratinocytes promotes wound healing in mice. J. Dermatol. Sci. 2017, 85, 106–114. [Google Scholar] [CrossRef]
- Rak, G.D.; Osborne, L.C.; Siracusa, M.C.; Kim, B.S.; Wang, K.; Bayat, A.; Artis, D.; Volk, S.W. IL-33-Dependent Group 2 Innate Lymphoid Cells Promote Cutaneous Wound Healing. J. Investig. Dermatol. 2016, 136, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Park, J.A.; Kim, J.; Rho, S.S.; Park, H.; Kim, Y.M.; Kwon, Y.G. Nuclear IL-33 is a transcriptional regulator of NF-κB p65 and induces endothelial cell activation. Biochem. Biophys. Res. Commun. 2012, 421, 305–311. [Google Scholar] [CrossRef]
- Strober, W.; Watanabe, T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease. Mucosal Immunol. 2011, 4, 484–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strober, W.; Murray, P.J.; Kitani, A.; Watanabe, T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 2006, 6, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strober, W.; Fuss, I.J. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Minaga, K.; Kamata, K.; Sakurai, T.; Komeda, Y.; Nagai, T.; Kitani, A.; Tajima, M.; Fuss, I.J.; Kudo, M.; et al. RICK/RIP2 is a NOD2-independent nodal point of gut inflammation. Int. Immunol. 2019, 31, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, M.; Watanabe, T.; Kamata, K.; Minaga, K.; Kudo, M. IL-33 as a Critical Cytokine for Inflammation and Fibrosis in Inflammatory Bowel Diseases and Pancreatitis. Front. Physiol. 2021, 12, 781012. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Műzes, G.; Molnár, B.; Tulassay, Z.; Sipos, F. Changes of the cytokine profile in inflammatory bowel diseases. World J. Gastroenterol. 2012, 18, 5848–5861. [Google Scholar] [CrossRef] [Green Version]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Parronchi, P.; Romagnani, P.; Annunziato, F.; Sampognaro, S.; Becchio, A.; Giannarini, L.; Maggi, E.; Pupilli, C.; Tonelli, F.; Romagnani, S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am. J. Pathol. 1997, 150, 823–832. [Google Scholar]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [Green Version]
- Kobori, A.; Yagi, Y.; Imaeda, H.; Ban, H.; Bamba, S.; Tsujikawa, T.; Saito, Y.; Fujiyama, Y.; Andoh, A. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J. Gastroenterol. 2010, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Sponheim, J.; Pollheimer, J.; Olsen, T.; Balogh, J.; Hammarström, C.; Loos, T.; Kasprzycka, M.; Sørensen, D.R.; Nilsen, H.R.; Küchler, A.M.; et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am. J. Pathol. 2010, 177, 2804–2815. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, M.D.; Goll, R.; Hol, J.; Olsen, T.; Rismo, R.; Sørbye, S.W.; Sundnes, O.; Haraldsen, G.; Florholmen, J. Loss of interleukin 33 expression in colonic crypts—A potential marker for disease remission in ulcerative colitis. Sci. Rep. 2016, 6, 35403. [Google Scholar] [CrossRef] [Green Version]
- Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Sedhom, M.A.; Pichery, M.; Murdoch, J.R.; Foligné, B.; Ortega, N.; Normand, S.; Mertz, K.; Sanmugalingam, D.; Brault, L.; Grandjean, T.; et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 2013, 62, 1714–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufford, M.M.; Kaplan, M.H. A gut reaction to IL-9. Nat. Immunol. 2014, 15, 599–600. [Google Scholar] [CrossRef] [Green Version]
- Oboki, K.; Ohno, T.; Kajiwara, N.; Arae, K.; Morita, H.; Ishii, A.; Nambu, A.; Abe, T.; Kiyonari, H.; Matsumoto, K.; et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 18581–18586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayamuro, H.; Yoshioka, Y.; Abe, Y.; Arita, S.; Katayama, K.; Nomura, T.; Yoshikawa, T.; Kubota-Koketsu, R.; Ikuta, K.; Okamoto, S.; et al. Interleukin-1 family cytokines as mucosal vaccine adjuvants for induction of protective immunity against influenza virus. J. Virol. 2010, 84, 12703–12712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikulski, Z.; Johnson, R.; Shaked, I.; Kim, G.; Nowyhed, H.; Goodman, W.; Chodaczek, G.; Pizarro, T.T.; Cominelli, F.; Ley, K. SAMP1/YitFc mice develop ileitis via loss of CCL21 and defects in dendritic cell migration. Gastroenterology 2015, 148, 783–793.e5. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Sharma, D.; Zhu, Q.; Karki, R.; Guy, C.S.; Vogel, P.; Kanneganti, T.D. IL-33 regulates the IgA-microbiota axis to restrain IL-1α-dependent colitis and tumorigenesis. J. Clin. Invest. 2016, 126, 4469–4481. [Google Scholar] [CrossRef] [PubMed]
- Tahaghoghi-Hajghorbani, S.; Ajami, A.; Ghorbanalipoor, S.; Hosseini-Khah, Z.; Taghiloo, S.; Khaje-Enayati, P.; Hosseini, V. Protective effect of TSLP and IL-33 cytokines in ulcerative colitis. Auto-Immun. Highlights 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Chen, J.; Xu, D.; Xie, Z.; Yu, B.; Tao, Y.; Shi, G.; Duan, L. IL-33-induced alternatively activated macrophage attenuates the development of TNBS-induced colitis. Oncotarget 2017, 8, 27704–27714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3⁺ regulatory T-cell responses in mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef]
- Holmén, N.; Lundgren, A.; Lundin, S.; Bergin, A.M.; Rudin, A.; Sjövall, H.; Ohman, L. Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm. Bowel. Dis. 2006, 12, 447–456. [Google Scholar] [CrossRef]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Groβ, P.; Doser, K.; Falk, W.; Obermeier, F.; Hofmann, C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm. Bowel. Dis. 2012, 18, 1900–1909. [Google Scholar] [CrossRef]
- Ngo Thi Phuong, N.; Palmieri, V.; Adamczyk, A.; Klopfleisch, R.; Langhorst, J.; Hansen, W.; Westendorf, A.M.; Pastille, E. IL-33 Drives Expansion of Type 2 Innate Lymphoid Cells and Regulatory T Cells and Protects Mice From Severe, Acute Colitis. Front. Immunol. 2021, 12, 669787. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- De Salvo, C.; Buela, K.A.; Creyns, B.; Corridoni, D.; Rana, N.; Wargo, H.L.; Cominelli, C.L.; Delaney, P.G.; Rodriguez-Palacios, A.; Cominelli, F.; et al. NOD2 drives early IL-33-dependent expansion of group 2 innate lymphoid cells during Crohn’s disease-like ileitis. J. Clin. Investig. 2021, 131, e140624. [Google Scholar] [CrossRef]
- Baumann, C.; Bonilla, W.V.; Fröhlich, A.; Helmstetter, C.; Peine, M.; Hegazy, A.N.; Pinschewer, D.D.; Löhning, M. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 4056–4061. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, W.V.; Fröhlich, A.; Senn, K.; Kallert, S.; Fernandez, M.; Johnson, S.; Kreutzfeldt, M.; Hegazy, A.N.; Schrick, C.; Fallon, P.G.; et al. The alarmin interleukin-33 drives protective antiviral CD8⁺ T cell responses. Science 2012, 335, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Bouma, G.; Strober, W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 2003, 3, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Hirai, F.; Andoh, A.; Ueno, F.; Watanabe, K.; Ohmiya, N.; Nakase, H.; Kato, S.; Esaki, M.; Endo, Y.; Yamamoto, H.; et al. Efficacy of Endoscopic Balloon Dilation for Small Bowel Strictures in Patients With Crohn’s Disease: A Nationwide, Multi-centre, Open-label, Prospective Cohort Study. J. Crohns. Colitis. 2018, 12, 394–401. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.R.; Bland, P.W.; Tarlton, J.F.; Peters, I.; Moorghen, M.; Sylvester, P.A.; Probert, C.S.; Whiting, C.V. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: A role for innate lymphoid cells? PLoS One 2012, 7, e52332. [Google Scholar] [CrossRef] [Green Version]
- Nishida, A.; Andoh, A.; Imaeda, H.; Inatomi, O.; Shiomi, H.; Fujiyama, Y. Expression of interleukin 1-like cytokine interleukin 33 and its receptor complex (ST2L and IL1RAcP) in human pancreatic myofibroblasts. Gut 2010, 59, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Camacho, K.; Diaz-Jimenez, D.; De la Fuente, M.; Quera, R.; Simian, D.; Martínez, M.; Landskron, G.; Olivares-Morales, M.; Cidlowski, J.A.; Xu, X.; et al. Inhibition of miR-378a-3p by Inflammation Enhances IL-33 Levels: A Novel Mechanism of Alarmin Modulation in Ulcerative Colitis. Front. Immunol. 2019, 10, 2449. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggeletopoulou, I.; Tsounis, E.P.; Triantos, C. Molecular Mechanisms Underlying IL-33-Mediated Inflammation in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 623. https://doi.org/10.3390/ijms24010623
Aggeletopoulou I, Tsounis EP, Triantos C. Molecular Mechanisms Underlying IL-33-Mediated Inflammation in Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2023; 24(1):623. https://doi.org/10.3390/ijms24010623
Chicago/Turabian StyleAggeletopoulou, Ioanna, Efthymios P. Tsounis, and Christos Triantos. 2023. "Molecular Mechanisms Underlying IL-33-Mediated Inflammation in Inflammatory Bowel Disease" International Journal of Molecular Sciences 24, no. 1: 623. https://doi.org/10.3390/ijms24010623
APA StyleAggeletopoulou, I., Tsounis, E. P., & Triantos, C. (2023). Molecular Mechanisms Underlying IL-33-Mediated Inflammation in Inflammatory Bowel Disease. International Journal of Molecular Sciences, 24(1), 623. https://doi.org/10.3390/ijms24010623