
The SDHD:p.H102R Variant Is Frequent in Russian Patients with Head and Neck Paragangliomas and Associated with Loss of 11p15.5 Region and Hypermethylation of H19-DMR
 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics and Identification of the SDHD:p.H102R Variant
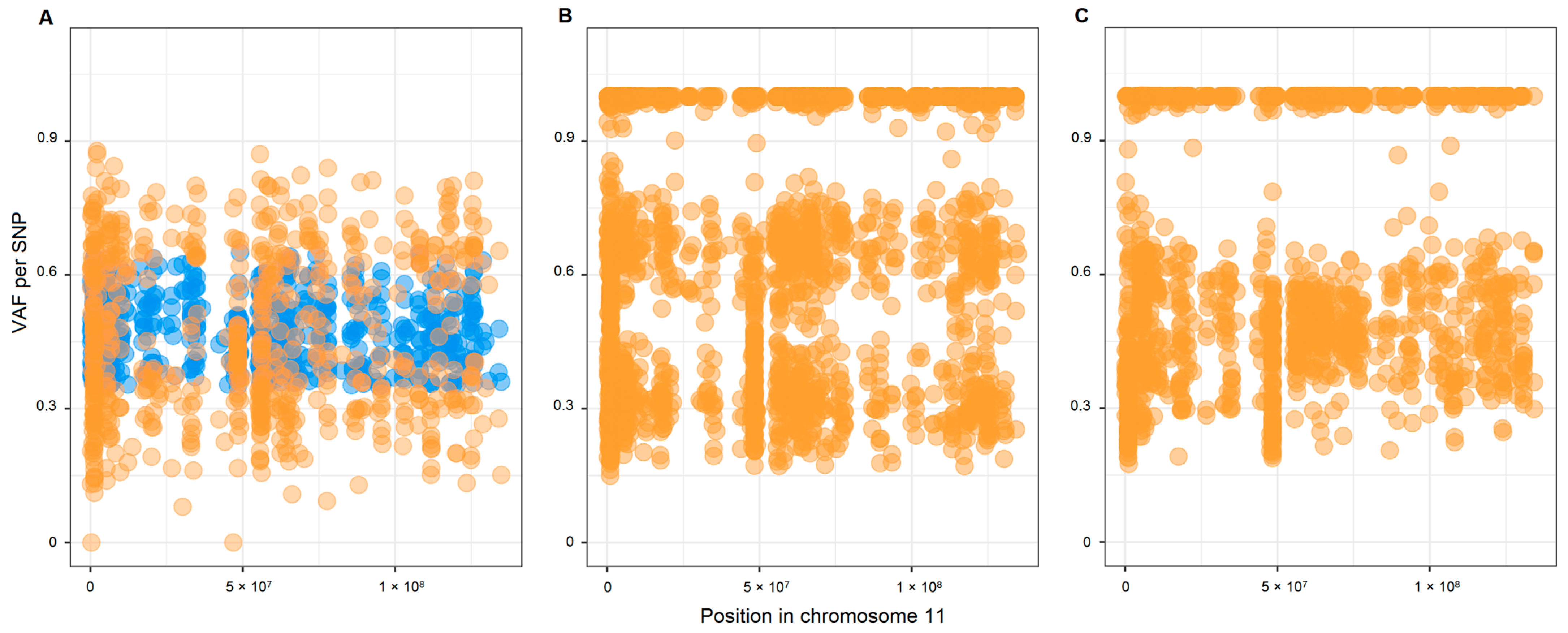
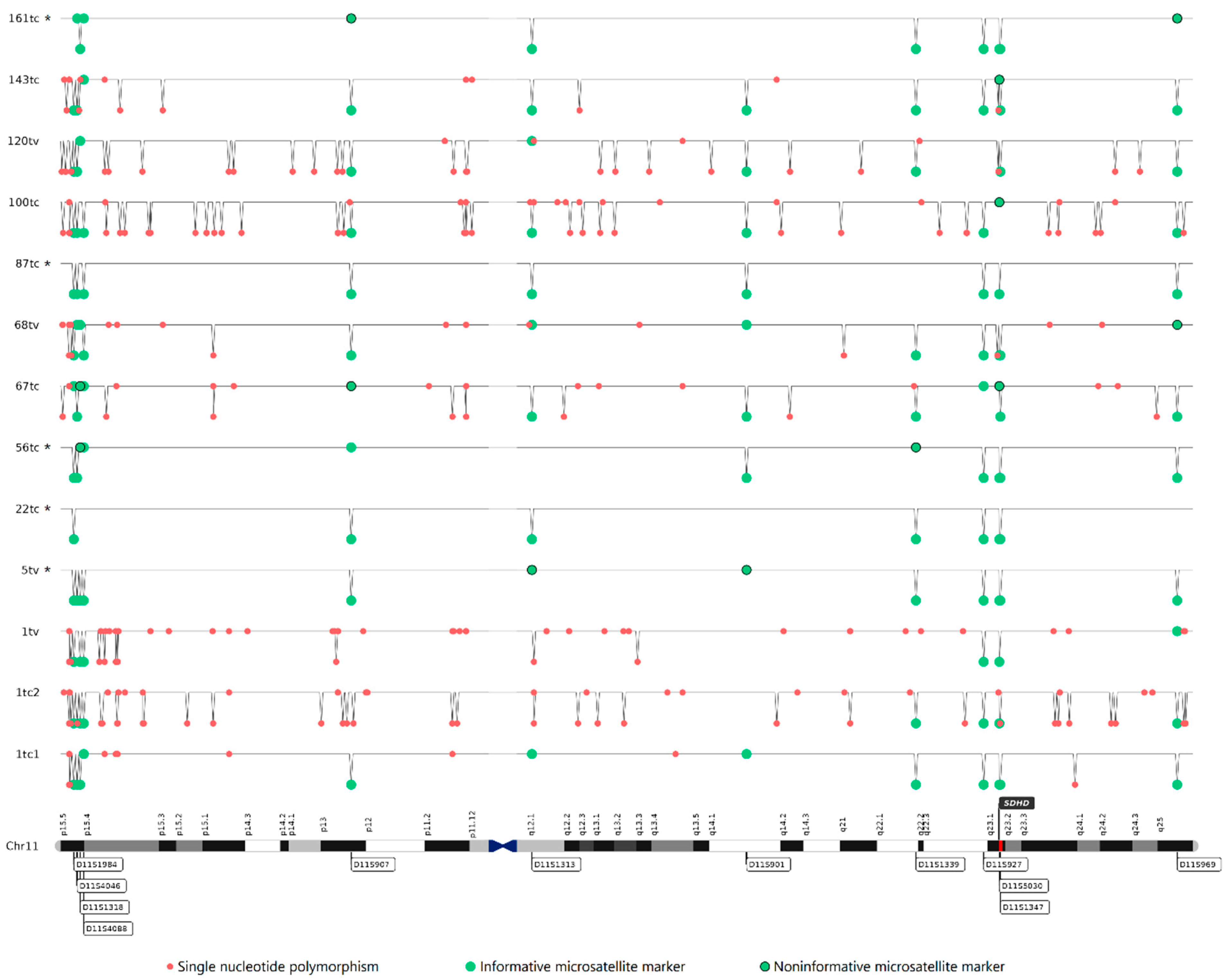
2.2. Copy Number Variations (CNVs) and Loss of Heterozygosity (LOH) in Chromosome 11
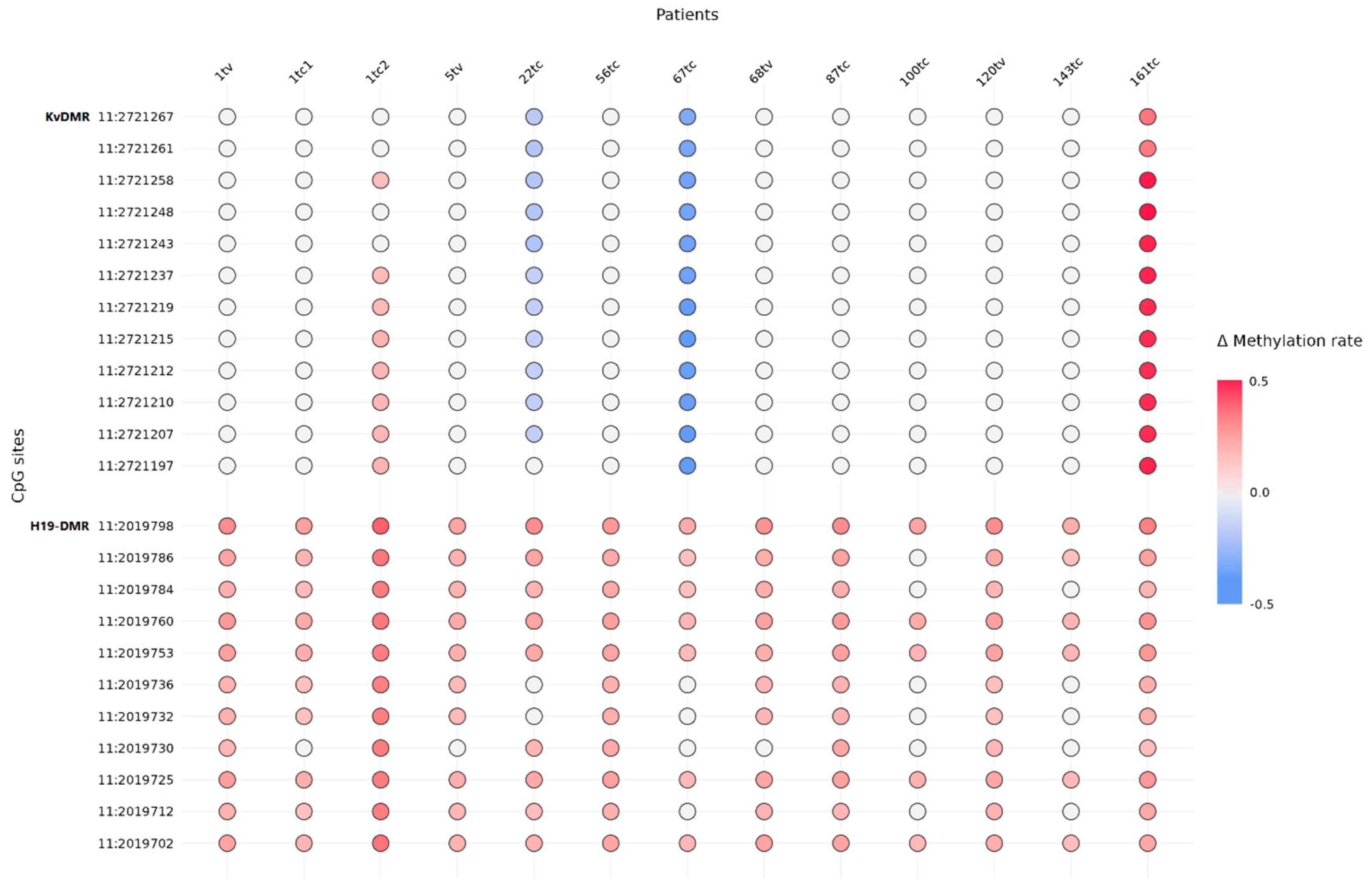
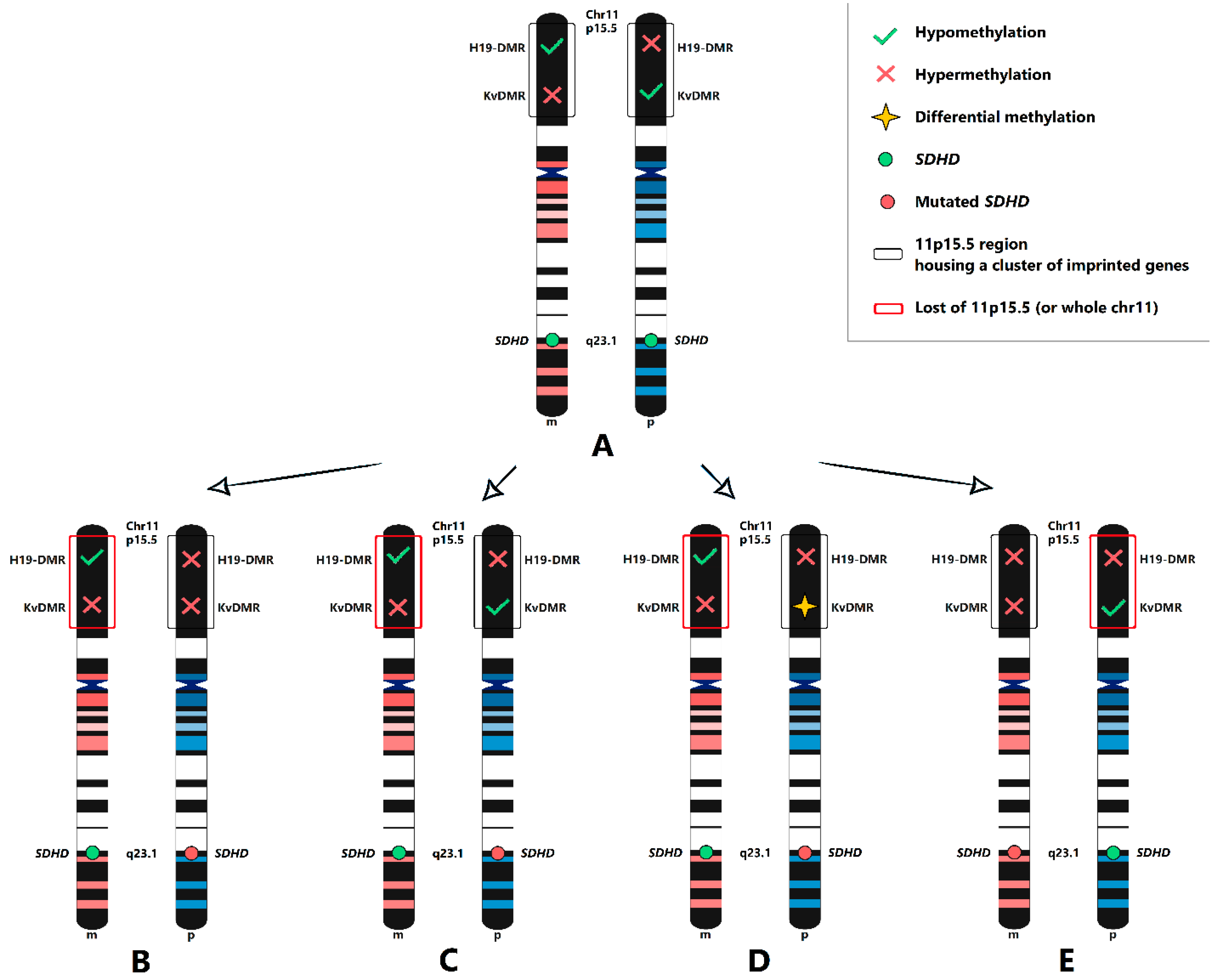
2.3. Maternal Allele Specific Loss of the 11p15.5 Region
2.4. SDH Complex Deficiency
3. Discussion
4. Materials and Methods
4.1. Human Tissues
4.2. DNA Isolation
4.3. Whole-Exome Sequencing of Case Cohort
4.4. Genetic Testing of Control Cohort
4.5. Copy Number Variation Analysis
4.6. Loss of Heterozygosity Analysis
4.7. Immunohistochemistry (IHC)
4.8. DNA Methylation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lloyd, R.; Osamura, R.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017.
- El-Naggar, A.K.; Chan, J.K.C.; Rubin Grandis, J.; Takata, T.; Slootweg, P.J.; International Agency for Research on C. WHO Classification of Head and Neck Tumours; WHO: Geneva, Switzerland, 2017.
- Snezhkina, A.; Pavlov, V.; Dmitriev, A.; Melnikova, N.; Kudryavtseva, A. Potential Biomarkers of Metastasizing Paragangliomas and Pheochromocytomas. Life 2021, 11, 1179. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1-5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baysal, B.E. Mitochondrial complex II and genomic imprinting in inheritance of paraganglioma tumors. Biochim. Biophys. Acta 2013, 1827, 573–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieldon, L.; William, D.; Hackmann, K.; Jahn, W.; Jahn, A.; Wagner, J.; Rump, A.; Bechmann, N.; Nolting, S.; Knosel, T.; et al. Optimizing Genetic Workup in Pheochromocytoma and Paraganglioma by Integrating Diagnostic and Research Approaches. Cancers 2019, 11, 809. [Google Scholar] [CrossRef] [Green Version]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Stenmark Askmalm, M.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef]
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle WA, USA, 1993. [Google Scholar]
- Snezhkina, A.V.; Fedorova, M.S.; Pavlov, V.S.; Kalinin, D.V.; Golovyuk, A.L.; Pudova, E.A.; Guvatova, Z.G.; Melnikova, N.V.; Dmitriev, A.A.; Razmakhaev, G.S.; et al. Mutation Frequency in Main Susceptibility Genes Among Patients With Head and Neck Paragangliomas. Front. Genet. 2020, 11, 614908. [Google Scholar] [CrossRef]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Hensen, E.F.; Jansen, J.C.; Siemers, M.D.; Oosterwijk, J.C.; Vriends, A.H.; Corssmit, E.P.; Bayley, J.P.; van der Mey, A.G.; Cornelisse, C.J.; Devilee, P. The Dutch founder mutation SDHD.D92Y shows a reduced penetrance for the development of paragangliomas in a large multigenerational family. Eur. J. Hum. Genet. 2010, 18, 62–66. [Google Scholar] [CrossRef]
- Pavlov, V.S.; Kalinin, D.V.; Lukyanova, E.N.; Golovyuk, A.L.; Fedorova, M.S.; Pudova, E.A.; Savvateeva, M.V.; Lipatova, A.V.; Guvatova, Z.G.; Kaprin, A.D.; et al. Multiple paragangliomas: A case report. BMC Med. Genom. 2020, 13, 125. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulskaya, M.V.; Shadrina, M.I.; Bakilina, N.A.; Zolotova, S.V.; Slominsky, P.A. The spectrum of SDHD mutations in Russian patients with head and neck paraganglioma. Int. J. Neurosci. 2018, 128, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Poeppel, T.D.; Yuece, A.; Boy, C.; Metz, K.A.; Kaminsky, E.; Neumann, H.P.; Rosenbaum, S.J.; Mann, K.; Moeller, L.C. Novel SDHD gene mutation (H102R) in a patient with metastatic cervical paraganglioma effectively treated by peptide receptor radionuclide therapy. J. Clin. Oncol. 2011, 29, e812–e815. [Google Scholar] [CrossRef] [PubMed]
- Taschner, P.E.; Jansen, J.C.; Baysal, B.E.; Bosch, A.; Rosenberg, E.H.; Brocker-Vriends, A.H.; van Der Mey, A.G.; van Ommen, G.J.; Cornelisse, C.J.; Devilee, P. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosom. Cancer 2001, 31, 274–281. [Google Scholar] [CrossRef] [PubMed]
- van Hulsteijn, L.T.; den Dulk, A.C.; Hes, F.J.; Bayley, J.P.; Jansen, J.C.; Corssmit, E.P. No difference in phenotype of the main Dutch SDHD founder mutations. Clin. Endocrinol. 2013, 79, 824–831. [Google Scholar] [CrossRef]
- Hensen, E.F.; van Duinen, N.; Jansen, J.C.; Corssmit, E.P.; Tops, C.M.; Romijn, J.A.; Vriends, A.H.; van der Mey, A.G.; Cornelisse, C.J.; Devilee, P.; et al. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin. Genet. 2012, 81, 284–288. [Google Scholar] [CrossRef]
- Simi, L.; Sestini, R.; Ferruzzi, P.; Gagliano, M.S.; Gensini, F.; Mascalchi, M.; Guerrini, L.; Pratesi, C.; Pinzani, P.; Nesi, G.; et al. Phenotype variability of neural crest derived tumours in six Italian families segregating the same founder SDHD mutation Q109X. J. Med. Genet. 2005, 42, e52. [Google Scholar] [CrossRef] [Green Version]
- Janecke, A.R.; Willett-Brozick, J.E.; Karas, C.; Hasipek, M.; Loeffler-Ragg, J.; Baysal, B.E. Identification of a 4.9-kilo base-pair Alu-mediated founder SDHD deletion in two extended paraganglioma families from Austria. J. Hum. Genet. 2010, 55, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Pasini, B.; Stratakis, C.A. SDH mutations in tumorigenesis and inherited endocrine tumours: Lesson from the phaeochromocytoma-paraganglioma syndromes. J. Intern. Med. 2009, 266, 19–42. [Google Scholar] [CrossRef] [Green Version]
- Heutink, P.; van der Mey, A.G.; Sandkuijl, L.A.; van Gils, A.P.; Bardoel, A.; Breedveld, G.J.; van Vliet, M.; van Ommen, G.J.; Cornelisse, C.J.; Oostra, B.A.; et al. A gene subject to genomic imprinting and responsible for hereditary paragangliomas maps to chromosome 11q23-qter. Hum. Mol. Genet. 1992, 1, 7–10. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensen, E.F.; Jordanova, E.S.; van Minderhout, I.J.; Hogendoorn, P.C.; Taschner, P.E.; van der Mey, A.G.; Devilee, P.; Cornelisse, C.J. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene 2004, 23, 4076–4083. [Google Scholar] [CrossRef] [Green Version]
- Pigny, P.; Vincent, A.; Cardot Bauters, C.; Bertrand, M.; de Montpreville, V.T.; Crepin, M.; Porchet, N.; Caron, P. Paraganglioma after maternal transmission of a succinate dehydrogenase gene mutation. J. Clin. Endocrinol. Metab. 2008, 93, 1609–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeap, P.M.; Tobias, E.S.; Mavraki, E.; Fletcher, A.; Bradshaw, N.; Freel, E.M.; Cooke, A.; Murday, V.A.; Davidson, H.R.; Perry, C.G.; et al. Molecular analysis of pheochromocytoma after maternal transmission of SDHD mutation elucidates mechanism of parent-of-origin effect. J. Clin. Endocrinol. Metab. 2011, 96, E2009–E2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayley, J.P.; Oldenburg, R.A.; Nuk, J.; Hoekstra, A.S.; van der Meer, C.A.; Korpershoek, E.; McGillivray, B.; Corssmit, E.P.; Dinjens, W.N.; de Krijger, R.R.; et al. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med. Genet. 2014, 15, 111. [Google Scholar] [CrossRef] [Green Version]
- Burnichon, N.; Mazzella, J.M.; Drui, D.; Amar, L.; Bertherat, J.; Coupier, I.; Delemer, B.; Guilhem, I.; Herman, P.; Kerlan, V.; et al. Risk assessment of maternally inherited SDHD paraganglioma and phaeochromocytoma. J. Med. Genet. 2017, 54, 125–133. [Google Scholar] [CrossRef]
- Hoekstra, A.S.; Hensen, E.F.; Jordanova, E.S.; Korpershoek, E.; van der Horst-Schrivers, A.N.; Cornelisse, C.; Corssmit, E.P.; Hes, F.J.; Jansen, J.C.; Kunst, H.P.; et al. Loss of maternal chromosome 11 is a signature event in SDHAF2, SDHD, and VHL-related paragangliomas, but less significant in SDHB-related paragangliomas. Oncotarget 2017, 8, 14525–14536. [Google Scholar] [CrossRef] [Green Version]
- Gill, A.J. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018, 72, 106–116. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kalinin, D.V.; Pavlov, V.S.; Lukyanova, E.N.; Golovyuk, A.L.; Fedorova, M.S.; Pudova, E.A.; Savvateeva, M.V.; Stepanov, O.A.; Poloznikov, A.A.; et al. Immunohistochemistry and Mutation Analysis of SDHx Genes in Carotid Paragangliomas. Int. J. Mol. Sci. 2020, 21, 6950. [Google Scholar] [CrossRef]
- Savvateeva, M.; Kudryavtseva, A.; Lukyanova, E.; Kobelyatskaya, A.; Pavlov, V.; Fedorova, M.; Pudova, E.; Guvatova, Z.; Kalinin, D.; Golovyuk, A.; et al. Somatic Mutation Profiling in Head and Neck Paragangliomas. J. Clin. Endocrinol. Metab. 2022, 107, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2018, 201178. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 18 July 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [Green Version]
- Masser, D.R.; Stanford, D.R.; Freeman, W.M. Targeted DNA methylation analysis by next-generation sequencing. J. Vis. Exp. JoVE 2015, 96, e52488. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Data | |
|---|---|---|
| Age, years | Min–max | 16–84 |
| Mean/median | 48.3/49 | |
| Sex, n (%) | Female | 101 (75%) |
| Male | 33 (25%) | |
| Tumor localization, n (%) | Carotid body * | 103 (77%) |
| Along the vagus nerve * | 33 (25%) | |
| N/A | 3 (2%) | |
| Tumor type, n (%) | Recurrent | 1 (0.7%) |
| Metastatic | 2 (1.3%) | |
| Multifocal | 10 (7.5%) | |
| Total, n | Patients | 134 |
| Tumors | 144 | |
| SDHx variant, n (%) | SDHA | 2 |
| SDHB | 15 | |
| SDHC | 5 | |
| SDHD | 35 | |
| Patient ID | Sex | Age, Year | Tumor Localization | SDHD:p.H102R Mutation Status | Variant in Other PPGL Susceptibility Genes | BAF-Analysis Result | SDHB IHC Staining |
|---|---|---|---|---|---|---|---|
| 1tc1 1tc2 1tv | F | 58 | Multifocal (bilateral carotid body tumor and unilateral vagal tumor) | Germline | - | Chr 11 − (all three tumors) | Weak diffuse (all three tumors) |
| 5tv | F | 61 | Vagal | Germline | - | Chr 11 − | Negative |
| 22tc | F | 63 | Carotid | Somatic | - | Chr 11 − | Weak diffuse |
| 56tc | F | 50 | Carotid | Germline | - | Chr 11 − | Weak diffuse |
| 67tc | F | 55 | Carotid | Germline | - | Chr 11 − | Positive |
| 68tv | F | 68 | Vagal | Germline | - | Chr 11 − | Weak diffuse |
| 87tc | F | 41 | Carotid | Germline | - | N/A | Positive |
| 100tc | F | 35 | Carotid | Germline | NF1—NM_000267: c.C5450G, p.S1817C (chr17:29654761, rs368654378) * | Chr 11 − | Weak diffuse |
| 120tv | F | 56 | Vagal | Germline | - | Chr 11 − | Positive |
| 135tc | F | 50 | Carotid | N/A | - | Chr 11 + | Positive |
| 143tc | F | 38 | Carotid | Germline | - | Chr 11 − | Heterogeneous (+/− regions) |
| 161tc | M | 38 | Carotid | Germline | - | N/A | Positive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snezhkina, A.; Fedorova, M.; Kobelyatskaya, A.; Markova, D.; Lantsova, M.; Ikonnikova, A.; Emelyanova, M.; Kalinin, D.; Pudova, E.; Melnikova, N.; et al. The SDHD:p.H102R Variant Is Frequent in Russian Patients with Head and Neck Paragangliomas and Associated with Loss of 11p15.5 Region and Hypermethylation of H19-DMR. Int. J. Mol. Sci. 2023, 24, 628. https://doi.org/10.3390/ijms24010628
Snezhkina A, Fedorova M, Kobelyatskaya A, Markova D, Lantsova M, Ikonnikova A, Emelyanova M, Kalinin D, Pudova E, Melnikova N, et al. The SDHD:p.H102R Variant Is Frequent in Russian Patients with Head and Neck Paragangliomas and Associated with Loss of 11p15.5 Region and Hypermethylation of H19-DMR. International Journal of Molecular Sciences. 2023; 24(1):628. https://doi.org/10.3390/ijms24010628
Chicago/Turabian StyleSnezhkina, Anastasiya, Maria Fedorova, Anastasiya Kobelyatskaya, Daria Markova, Margarita Lantsova, Anna Ikonnikova, Marina Emelyanova, Dmitry Kalinin, Elena Pudova, Nataliya Melnikova, and et al. 2023. "The SDHD:p.H102R Variant Is Frequent in Russian Patients with Head and Neck Paragangliomas and Associated with Loss of 11p15.5 Region and Hypermethylation of H19-DMR" International Journal of Molecular Sciences 24, no. 1: 628. https://doi.org/10.3390/ijms24010628