
Drug Repurposing against KRAS Mutant G12C: A Machine Learning, Molecular Docking, and Molecular Dynamics Study



Abstract
:1. Introduction
2. Results and Discussion
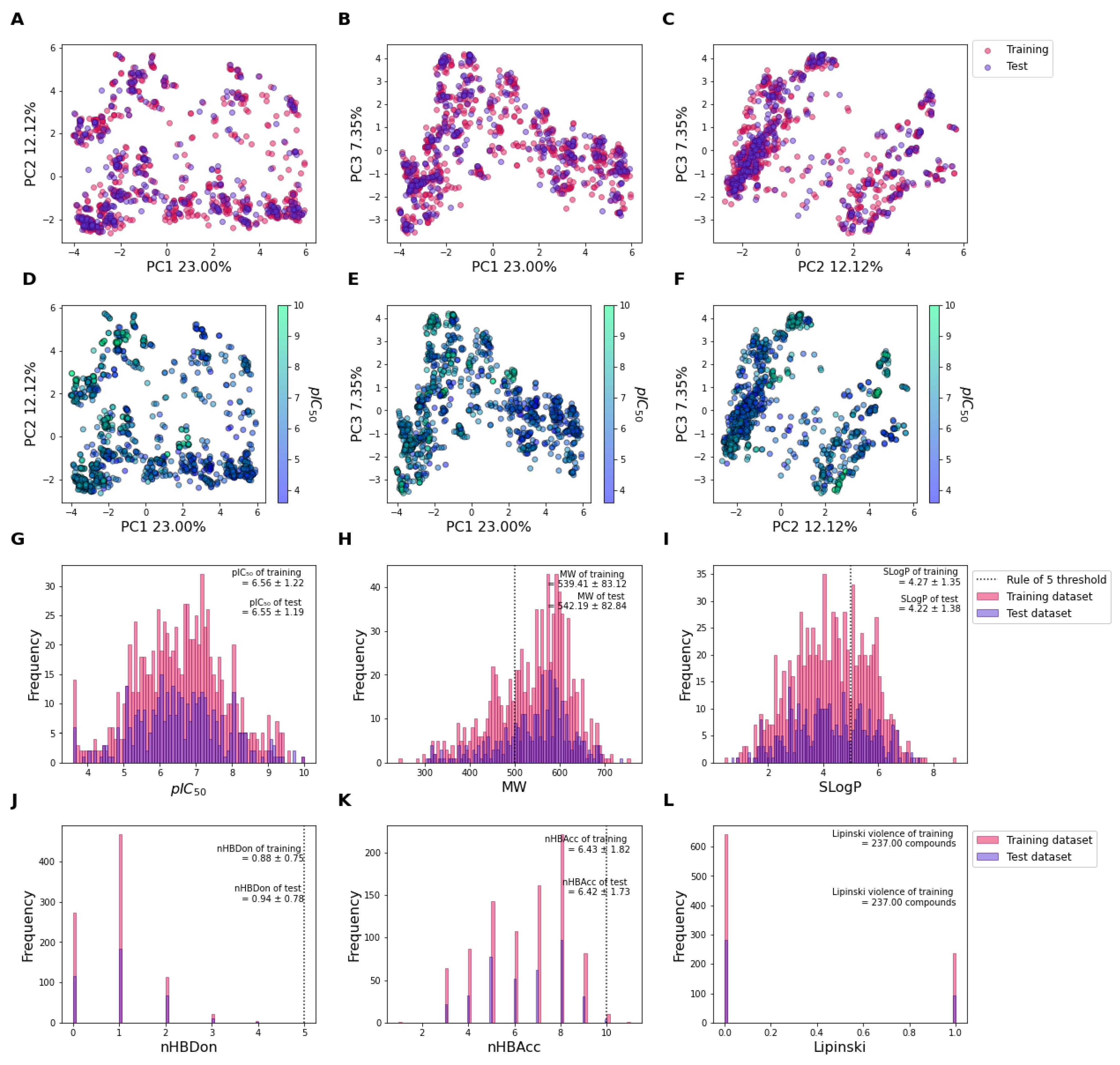
2.1. Chemical Distribution of the Datasets
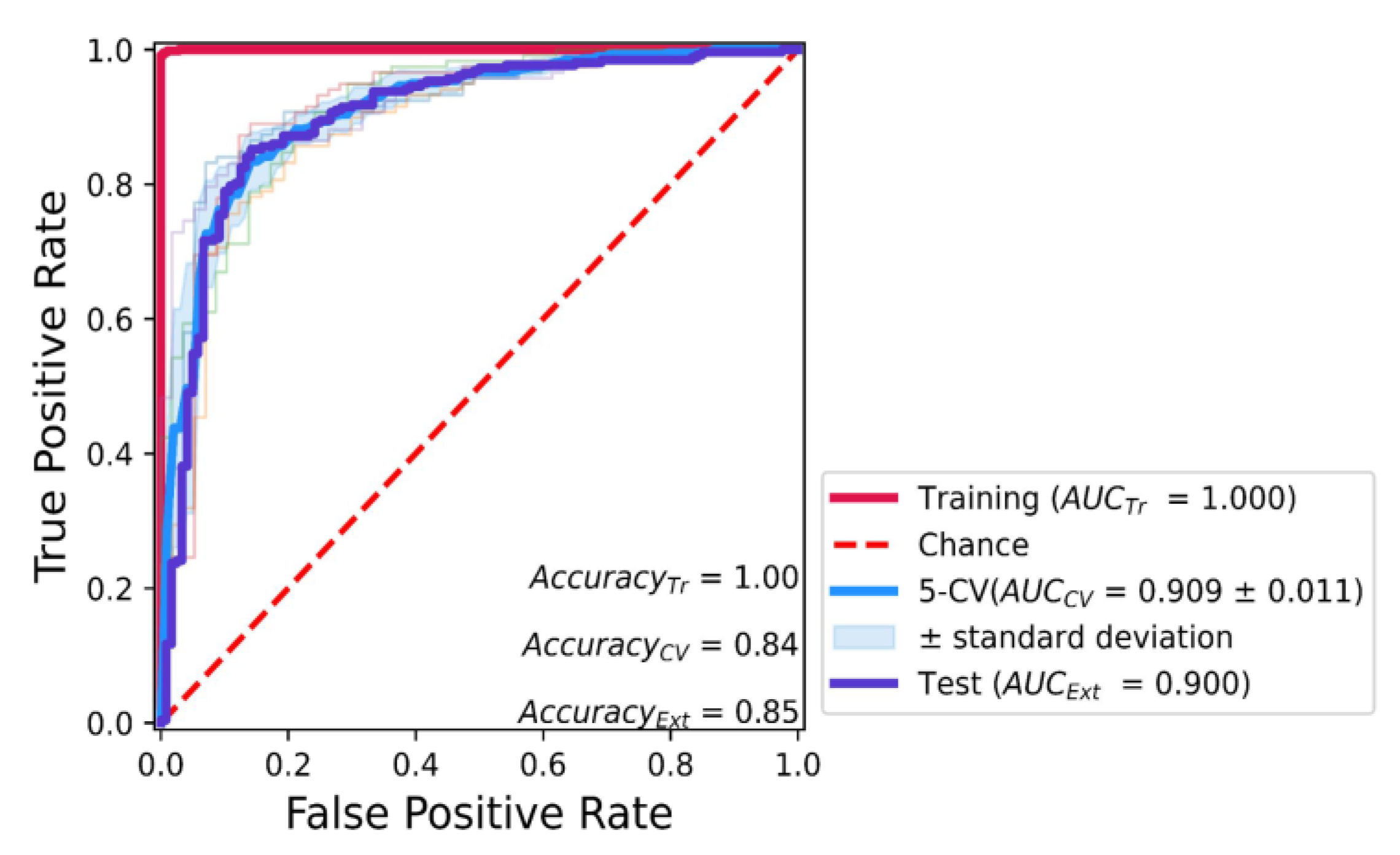
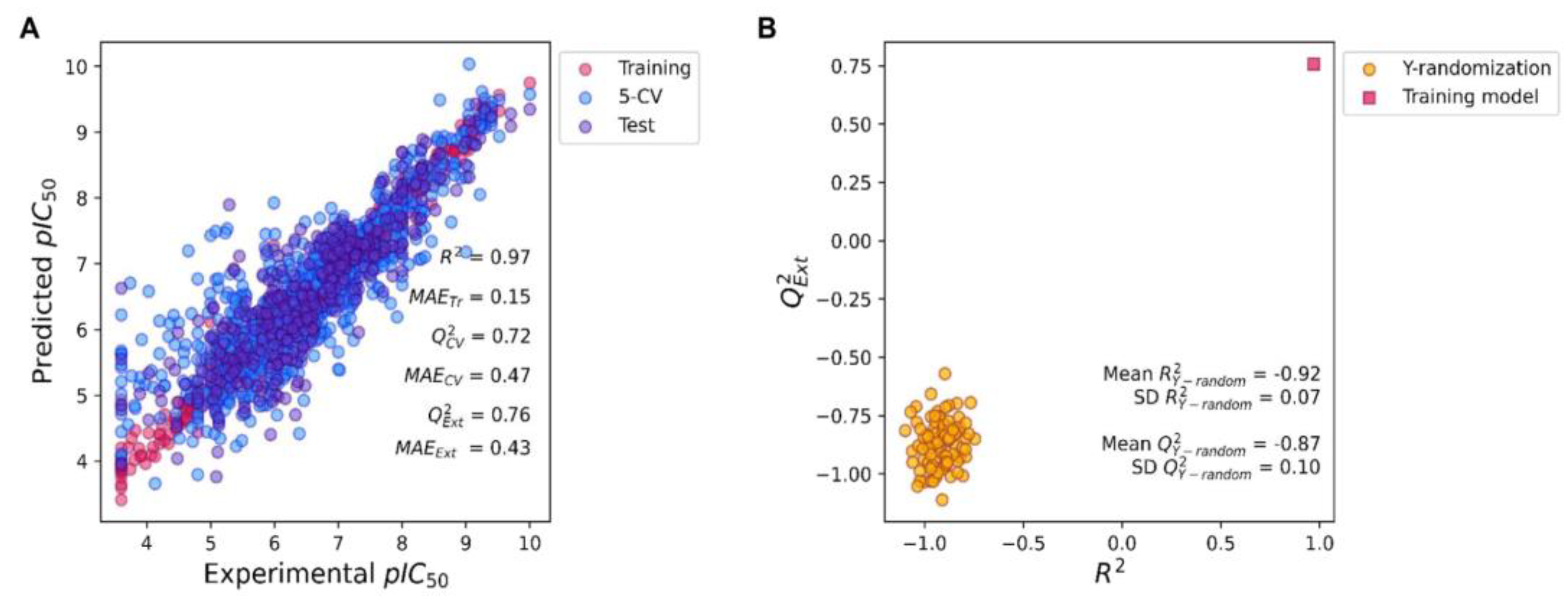
2.2. Predictive KRAS pIC50 ML Model Based on XGBoost Algorithm of All Molecular Fingerprints
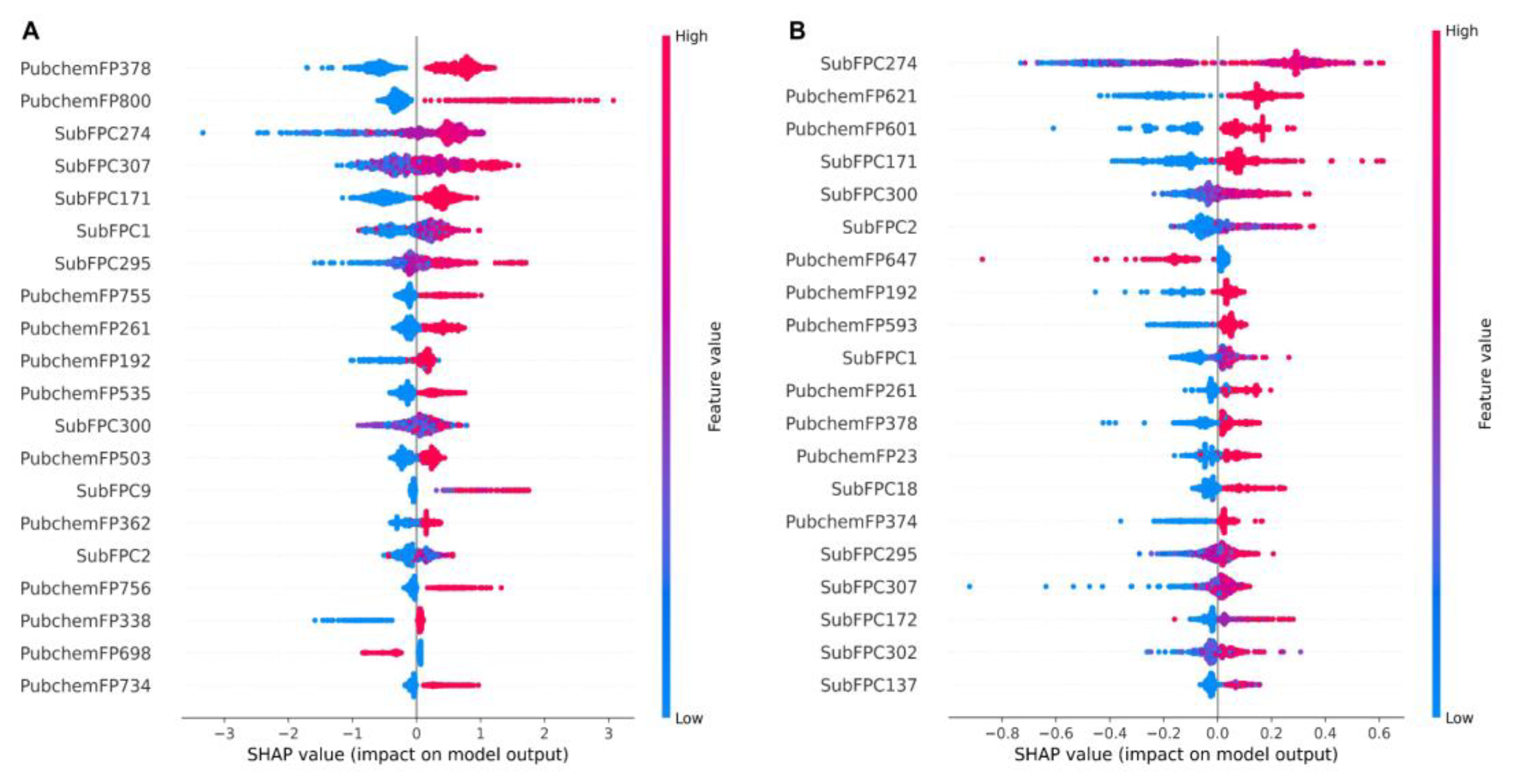
2.3. KRAS Inhibitor Structural Importance Identified by SHAP Algorithm
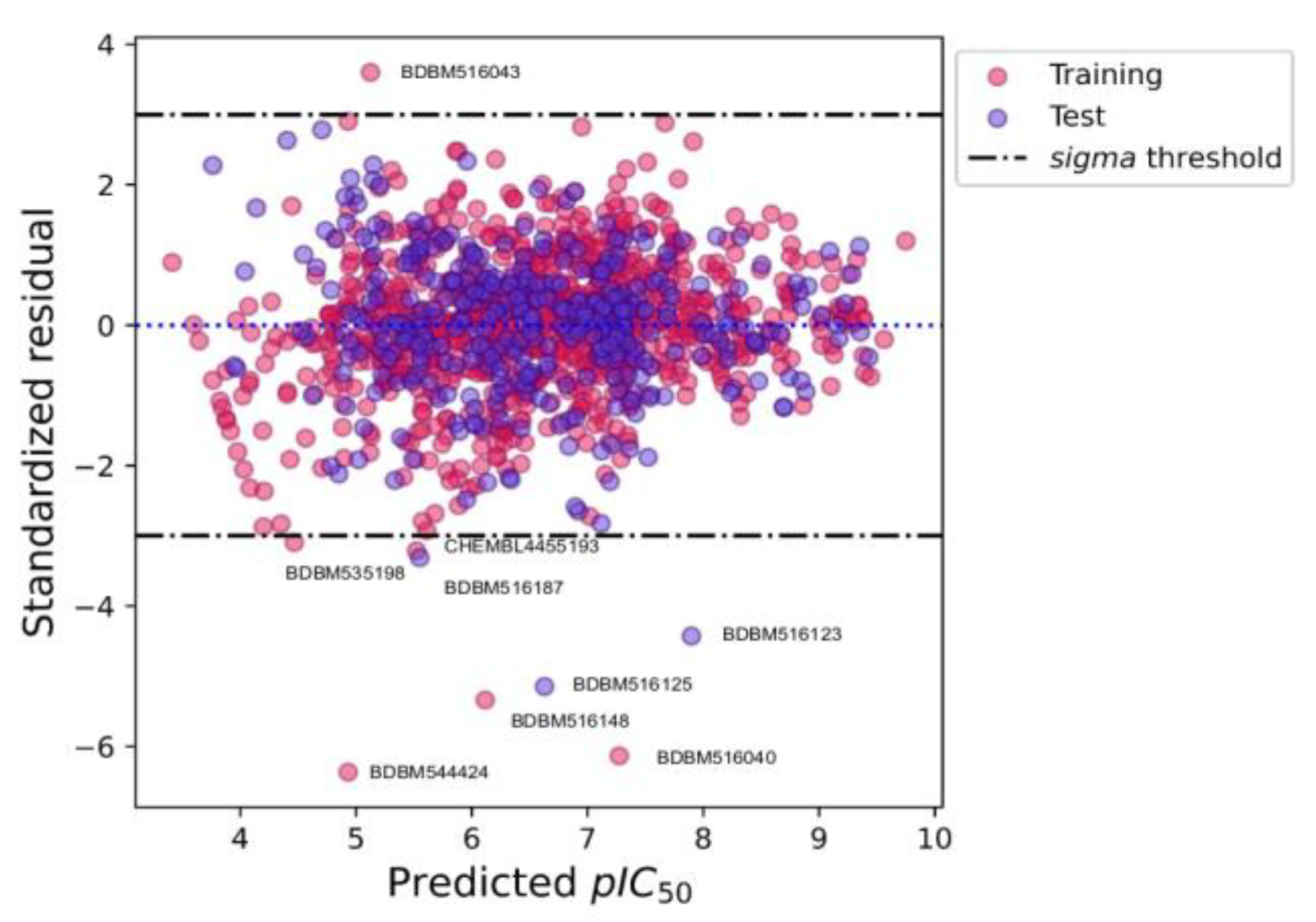
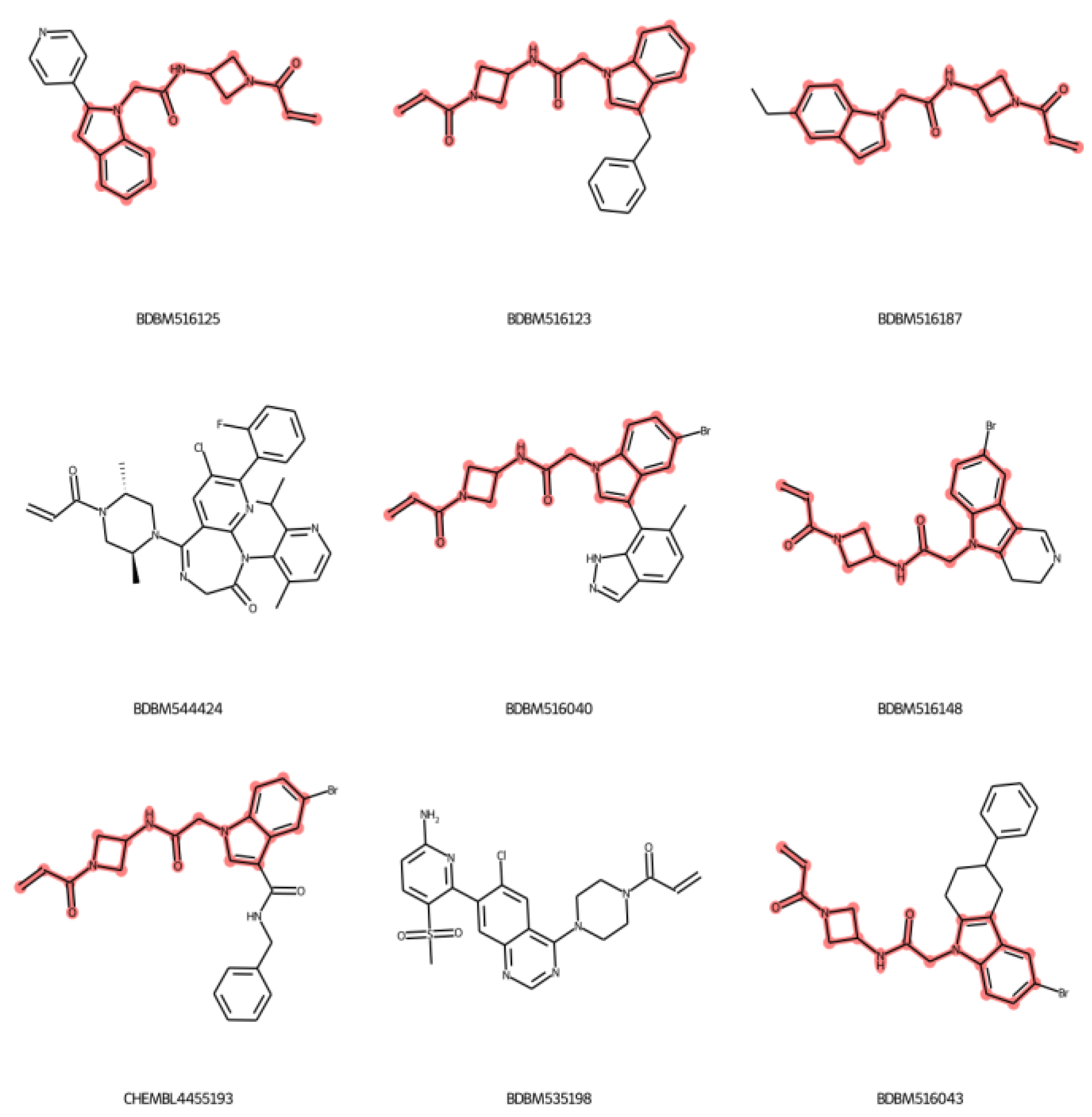
2.4. Applicability Domain
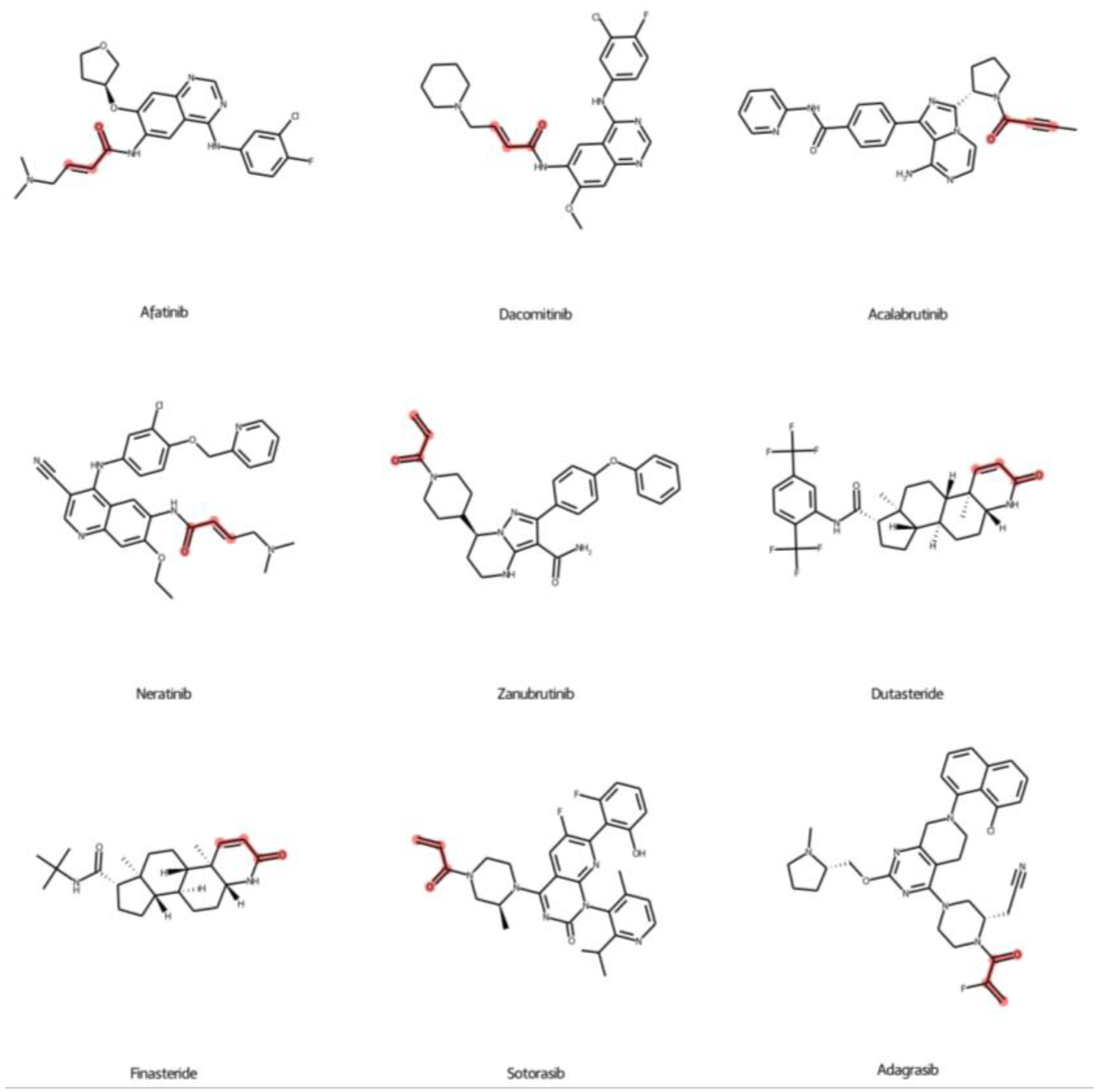
2.5. Prediction of FDA-Approved Drugs against KRASG12C Protein
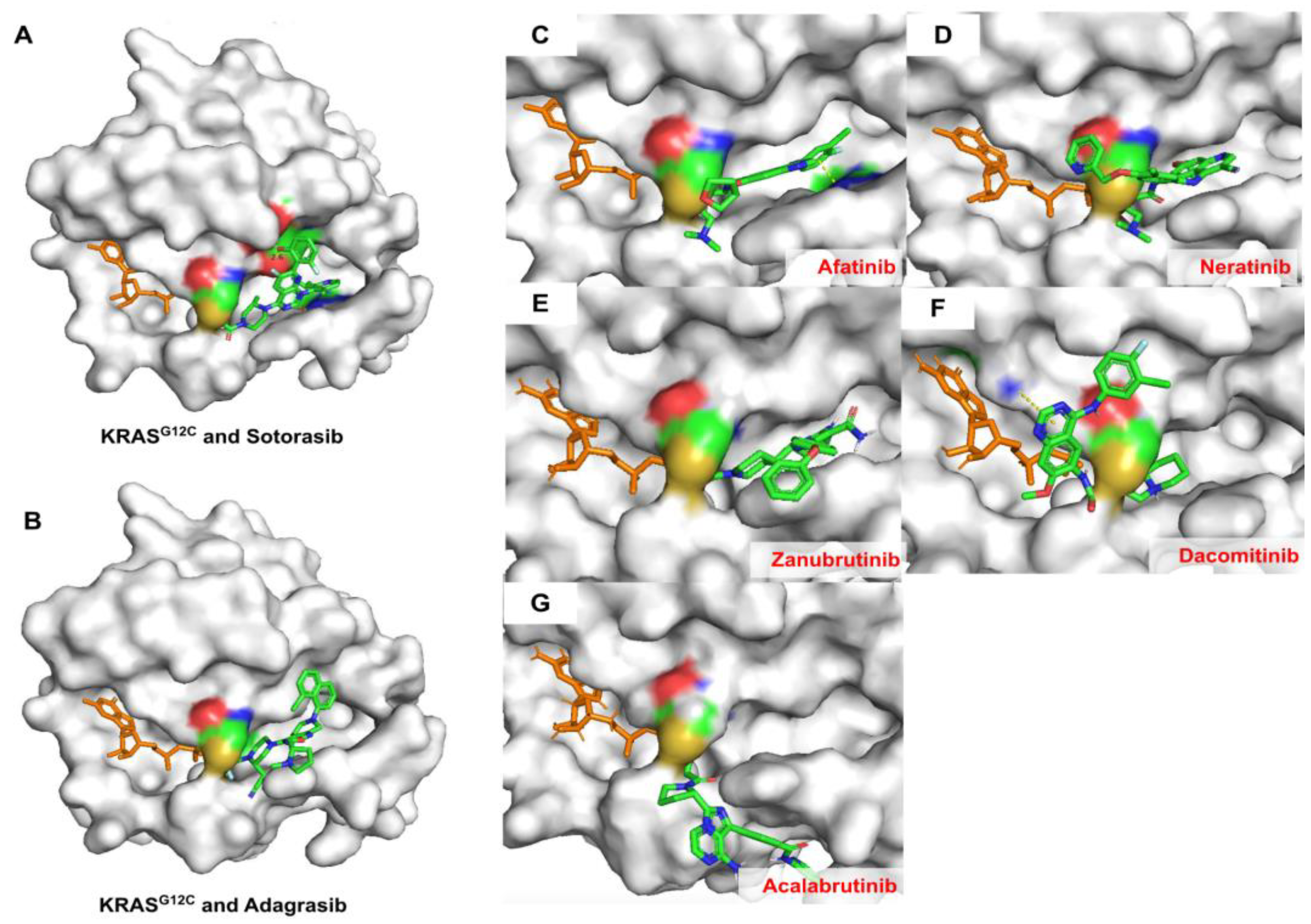
2.6. Covalent Docking of Predicted Compounds against KRASG12C Protein
2.7. Molecular Dynamics Simulation of FDA-Approved Drugs and KRAS G12C Protein
2.8. Summary of New Predicted FDA-Approved Drugs against KRAS Mutations
3. Materials and Methods
3.1. Data Collection
3.2. Molecular Fingerprints and Molecular Descriptors Calculation
3.3. Extreme Gradient Boosting
3.4. Classification Model Construction
3.5. Classification Model Evaluation
3.6. Regression Model Construction
3.7. Regression Model Evaluation
3.8. Y-Randomization
3.9. Feature Importance
3.10. Applicability Domain
3.11. Covalent Docking
3.12. Molecular Dynamic Simulation
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, J.; Zang, Q.; Wen, Y.; Pan, Z.; Yao, Z.; Huang, M.; Huang, J.; Chen, J.; Wang, R. Prognostic Value of KRAS Mutation in Patients Undergoing Pulmonary Metastasectomy for Colorectal Cancer: A Systematic Review and Meta-Analysis. Crit. Rev. Oncol./Hematol. 2021, 160, 103308. [Google Scholar] [CrossRef] [PubMed]
- Formica, V.; Sera, F.; Cremolini, C.; Riondino, S.; Morelli, C.; Arkenau, H.-T.; Roselli, M. KRAS and BRAF Mutations in Stage II and III Colon Cancer: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 2022, 114, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.P.; Sutton, P.A.; Evans, J.P.; Clifford, R.; McAvoy, A.; Lewis, J.; Rousseau, A.; Mountford, R.; McWhirter, D.; Malik, H.Z. Specific Mutations in KRAS Codon 12 Are Associated with Worse Overall Survival in Patients with Advanced and Recurrent Colorectal Cancer. Br. J. Cancer 2017, 116, 923–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, G.; Khurshid, F.; Lu, K.; Woodward, B.; Husain, H. Selective KRAS G12C Inhibitors in Non-Small Cell Lung Cancer: Chemistry, Concurrent Pathway Alterations, and Clinical Outcomes. NPJ Precis. Oncol. 2021, 5, 98. [Google Scholar] [CrossRef]
- Terrell, E.M. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 2019, 76, 872–884. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS G12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the Undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight Toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non–Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C Inhibitor: Activity and Resistance. Cancer Gene Ther. 2022, 29, 875–878. [Google Scholar] [CrossRef]
- Rodrigues, R.; Duarte, D.; Vale, N. Drug Repurposing in Cancer Therapy: Influence of Patient’s Genetic Background in Breast Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 4280. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From Angina to Erectile Dysfunction to Pulmonary Hypertension and Beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an Adjuvant Treatment for Cancer: A Systematic Review and Meta-Analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- El Zarif, T.; Yibirin, M.; De Oliveira-Gomes, D.; Machaalani, M.; Nawfal, R.; Bittar, G.; Bahmad, H.F.; Bitar, N. Overcoming Therapy Resistance in Colon Cancer by Drug Repurposing. Cancers 2022, 14, 2105. [Google Scholar] [CrossRef]
- Bioinformatics: Methods and Applications, 1st ed.; Singh, D.B.; Pathak, P.K. (Eds.) Elsevier: Waltham, MA, USA, 2021; ISBN 978-0-323-89775-4. [Google Scholar]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and Deep Learning Approaches for Cancer Drug Repurposing. Semin. Cancer Biol. 2021, 68, 132–142. [Google Scholar] [CrossRef]
- Oyedele, A.-Q.K.; Ogunlana, A.T.; Boyenle, I.D.; Ibrahim, N.O.; Gbadebo, I.O.; Owolabi, N.A.; Ayoola, A.M.; Francis, A.C.; Eyinade, O.H.; Adelusi, T.I. Pharmacophoric Analogs of Sotorasib-Entrapped KRAS G12C in Its Inactive GDP-Bound Conformation: Covalent Docking and Molecular Dynamics Investigations. Mol. Divers. 2022. [Google Scholar] [CrossRef]
- Anwaar, M.U.; Adnan, F.; Abro, A.; Khan, R.A.; Rehman, A.U.; Osama, M.; Rainville, C.; Kumar, S.; Sterner, D.E.; Javed, S.; et al. Combined Deep Learning and Molecular Docking Simulations Approach Identifies Potentially Effective FDA Approved Drugs for Repurposing against SARS-CoV-2. Comput. Biol. Med. 2021, 141, 105049. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Gao, J.; Weng, G.; Ding, J.; Chai, X.; Pang, J.; Kang, Y.; Li, D.; Cao, D.; Hou, T. CovalentInDB: A Comprehensive Database Facilitating the Discovery of Covalent Inhibitors. Nucleic Acids Res. 2021, 49, D1122–D1129. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.P.; Wang, W.M.; Liaw, A.; Ma, J.; Gifford, E.M. Extreme Gradient Boosting as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2016, 56, 2353–2360. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A Public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S.-I. From Local Explanations to Global Understanding with Explainable AI for Trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef]
- OECD Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Model. Available online: https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2007)2 (accessed on 25 August 2022).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Bray, S.A.; Senapathi, T.; Barnett, C.B.; Grüning, B.A. Intuitive, Reproducible High-Throughput Molecular Dynamics in Galaxy: A Tutorial. J. Cheminform. 2020, 12, 54. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-Small Cell Lung Cancer: Current Treatment and Future Advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- International Human Genome Sequencing Consortium; Whitehead Institute for Biomedical Research, Center for Genome Research; Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, V.; Ziehr, J.; Agulnik, M.; Johnson, M. Afatinib: Emerging Next-Generation Tyrosine Kinase Inhibitor for NSCLC. OncoTargets Ther. 2013, 2013, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Moll, H.P.; Pranz, K.; Musteanu, M.; Grabner, B.; Hruschka, N.; Mohrherr, J.; Aigner, P.; Stiedl, P.; Brcic, L.; Laszlo, V.; et al. Afatinib Restrains K-RAS–Driven Lung Tumorigenesis. Sci. Transl. Med. 2018, 10, eaao2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, E.D. Neratinib: First Global Approval. Drugs 2017, 77, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Moy, B.; Mansi, J.; Ejlertsen, B.; Holmes, F.A.; Chia, S.; Iwata, H.; Gnant, M.; Loibl, S.; Barrios, C.H.; et al. Final Efficacy Results of Neratinib in HER2-Positive Hormone Receptor-Positive Early-Stage Breast Cancer from the Phase III ExteNET Trial. Clin. Breast Cancer 2021, 21, 80–91.e7. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Booth, L.; Roberts, J.L.; Liu, J.; Poklepovic, A.; Lalani, A.S.; Tuveson, D.; Martinez, J.; Hancock, J.F. Neratinib Inhibits Hippo/YAP Signaling, Reduces Mutant K-RAS Expression, and Kills Pancreatic and Blood Cancer Cells. Oncogene 2019, 38, 5890–5904. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Sander, C.; Lalani, A.S.; Kirkwood, J.M.; Hancock, J.F.; Poklepovic, A.; Dent, P. Neratinib and Entinostat Combine to Rapidly Reduce the Expression of K-RAS, N-RAS, Gαq and Gα11 and Kill Uveal Melanoma Cells. Cancer Biol. Ther. 2019, 20, 700–710. [Google Scholar] [CrossRef]
- Dent, P.; Booth, L.; Poklepovic, A.; Von Hoff, D.; Martinez, J.; Zhou, Y.; Hancock, J.F. Osimertinib-Resistant NSCLC Cells Activate ERBB2 and YAP/TAZ and Are Killed by Neratinib. Biochem. Pharmacol. 2021, 190, 114642. [Google Scholar] [CrossRef]
- Dent, P.; Booth, L.; Poklepovic, A.; Hancock, J.F. Neratinib as a Potential Therapeutic for Mutant RAS and Osimertinib-Resistant Tumours. EMJ 2022, 7, 127–133. [Google Scholar] [CrossRef]
- Li, G.; Liu, X.; Chen, X. Simultaneous Development of Zanubrutinib in the USA and China. Nat. Rev. Clin. Oncol. 2020, 17, 589–590. [Google Scholar] [CrossRef]
- Čermáková, L.; Hofman, J.; Laštovičková, L.; Havlíčková, L.; Špringrová, I.; Novotná, E.; Wsól, V. Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux. Pharmaceutics 2022, 14, 1994. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Autin, L.; Fuentes, D.; Maritan, M.; Barad, B.A.; Medina, M.; Olson, A.J.; Grotjahn, D.A.; Goodsell, D.S. CellPAINT: Turnkey Illustration of Molecular Cell Biology. Front. Bioinform. 2021, 1, 660936. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-Descriptor: An Open Source Software to Calculate Molecular Descriptors and Fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.-S.; Kawashita, N.; Takagi, T. Mordred: A Molecular Descriptor Calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 All-Atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Fingerprints | Descriptions | Examples 1 |
|---|---|---|
| PubChemFP378 | C(~N)(:C)(:N) |  |
| SubFPC274 | Aromatic atom | 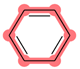 |
| SubFPC307 | Chiral center | 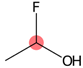 |
| SubFPC171 | Aryl Chloride | 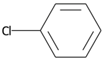 |
| SubFPC1 | Primary carbon |  |
| SubFPC295 | C-O, N, or S bond | 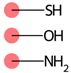 |
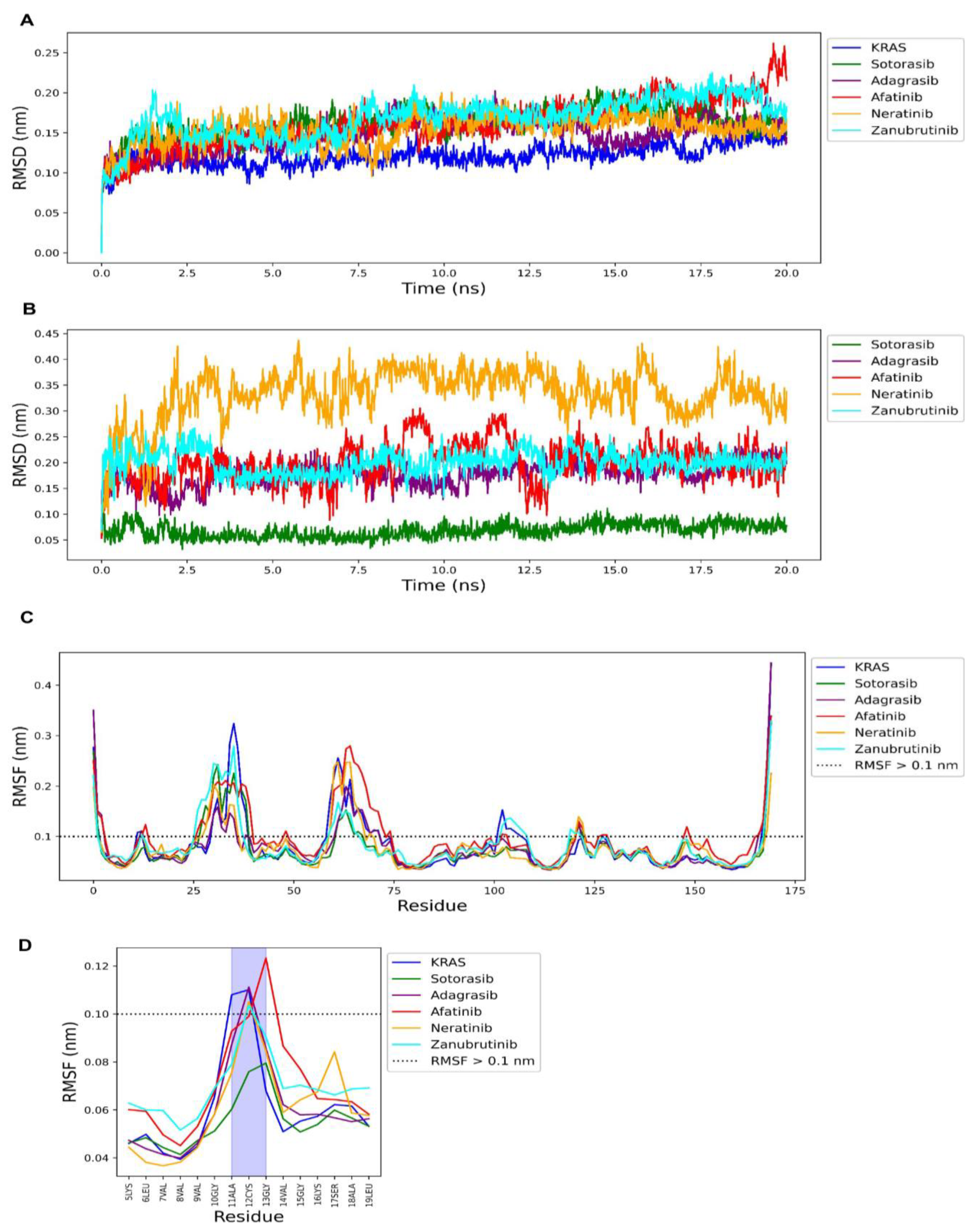
| PubChemFP261 | ≥4 aromatic rings | 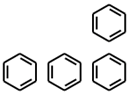 |
| PubChemFP192 | ≥3 any ring size 6 | 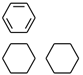 |
| SubFPC300 | 1,3-tautomerizable | 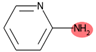 |
| SubFPC2 | Secondary carbon |  |
| Name | Predicted Classes | Predicted pIC50 | Experimental pIC50 † | Primary Targets [30] | Primary Indications [30] |
|---|---|---|---|---|---|
| Afatinib | Active | 7.43 | No report | EGFR and HER2 | NSCLC |
| Dacomitinib | Active | 7.32 | No report | EGFR T790M | NSCLC |
| Acalabrutinib | Active | 6.75 | No report | BTK | MCL |
| Neratinib | Active | 6.68 | No report | EGFR, HER2, HER4 | Breast cancer |
| Zanubrutinib | Active | 6.49 | No report | BTK | MCL |
| Dutasteride | Active | 5.94 | No report | 5α-Reductase | BPH |
| Finasteride | Active | 5.05 | No report | 5α-Reductase | Alopecia, BPH |
| Sotorasib * | Active | 6.95 | 7.52 | KRASG12C | NSCLC |
| Adagrasib * | Active | 8.02 | 8.30 | KRASG12C | NSCLC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srisongkram, T.; Weerapreeyakul, N. Drug Repurposing against KRAS Mutant G12C: A Machine Learning, Molecular Docking, and Molecular Dynamics Study. Int. J. Mol. Sci. 2023, 24, 669. https://doi.org/10.3390/ijms24010669
Srisongkram T, Weerapreeyakul N. Drug Repurposing against KRAS Mutant G12C: A Machine Learning, Molecular Docking, and Molecular Dynamics Study. International Journal of Molecular Sciences. 2023; 24(1):669. https://doi.org/10.3390/ijms24010669
Chicago/Turabian StyleSrisongkram, Tarapong, and Natthida Weerapreeyakul. 2023. "Drug Repurposing against KRAS Mutant G12C: A Machine Learning, Molecular Docking, and Molecular Dynamics Study" International Journal of Molecular Sciences 24, no. 1: 669. https://doi.org/10.3390/ijms24010669